Tumour heterogeneity is a phenomenon where the cancer cells evolve diversely over the course of the disease. As a result of the evolution, the cancer cells can be found to be genetically, epigenetically and/or phenotypically different in order to survive in the human body. The tumour microenvironment also plays a crucial role during the evolution.
- melanoma
- tumour heterogeneity
- intertumoural heterogeneity
1. Introduction
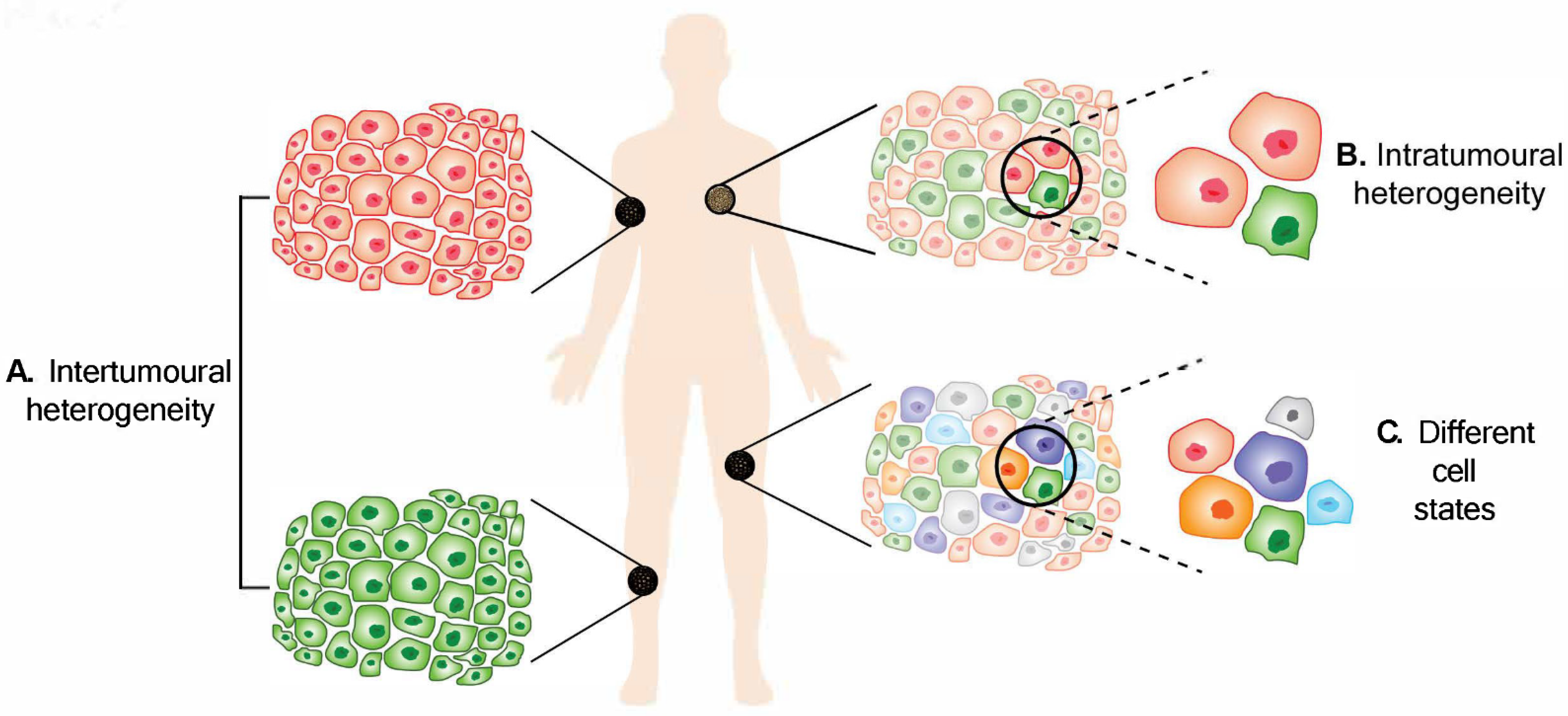
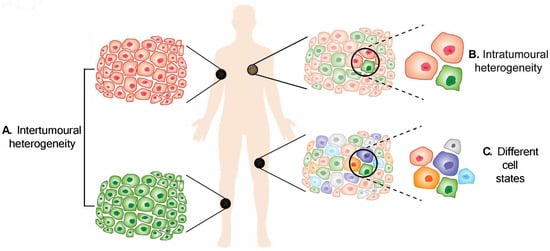
Melanoma heterogeneity. (
) Intertumoural heterogeneity refers to differences between lesions in a patient, while (
) intratumoural heterogeneity describes the differences between cells or subpopulations of cells within a tumour. Advancement in technology has revealed (
) different cell states in melanoma tumours.
2. Intertumoural Heterogeneity
3. Intratumoural Heterogeneity
Intratumoural heterogeneity describes the differences between cells or subpopulations of cells within a tumour. Heterogeneous cells within a tumour can display remarkable variability in phenotypic traits (Table 1). A cross-sectional analysis that screened 1165 tumour exomes from 12 different cancers revealed that intratumoural heterogeneity was found in all cancer types, with 86% of the samples consisting of at least two subpopulations, with melanoma being the most heterogeneous [18]. Furthermore, a study has shown that 6/9 (67%) melanoma tumours exhibited intratumoural heterogeneity [25].
3.1. Genetic Contribution to Intratumoural Heterogeneity
3.2. Intratumoural Heterogeneity from Non-Genetic Sources
3.2.1. Epigenetic Heterogeneity
3.2.2. Phenotypic Heterogeneity
Tumour Microenvironment
Immune Heterogeneity
4. Impact of Heterogeneity on Treatment Responses
The overall survival rate for metastatic melanoma patients has significantly improved owing to the development and advancement in targeted therapies and immunotherapies [120][103]. However, there is a large proportion of patients who do not benefit from these new treatments due to differential responses resulting from tumour heterogeneity. Resistance to BRAFi treatment is often associated with reactivation of the MAPK pathway through mechanisms such as RAS mutation, BRAF amplification and alternative splicing [121][104]. For example, a patient developed resistance to BRAFi treatment after subpopulations in the tumour switched from NRASWT/BRAFV600E to NRASG13R/BRAFV600E, thereby reactivating the MAPK pathway [44][42]. In a rare case of transplacental transmission of metastatic melanoma with BRAFV600E mutation, the mother was refractory to BRAFi treatment, whereas the baby was responsive to therapy [122][105]. Genomic analyses showed that there were at least two related but distinct subpopulations causing resistance, which later led to relapse. Notably, single-cell analysis of a panel of 16 melanoma cell lines and a primary cell line (epidermal melanocyte) found the status of markers, such as MITF, AXL and NGFR, impacted the BRAF/MEKi treatment [123][106]. AXLhigh cells were more resistant to various treatments in comparison to MITFhigh or NGFRhigh cells. Resistant AXLhigh cells exhibited partial MAPK pathway inhibition with high Ki67 expression, while both MITFhigh and NGFRhigh cells demonstrated considerable inhibition of the MAPK pathway, suggesting that the MAPK pathway is crucial for AXLhigh cells but not MITFhigh and NGFRhigh cells. Co-treatment of BRAF/MEKi with different epigenetic modulators in the cells revealed selective sensitivities presented by different populations of cells [123][106]. Several slow-cycling subpopulations, such as MITFlow/NF-κBhigh/AXLhigh, are also correlated to reactivation of the MAPK pathway [68,69][75][76]. Importantly, elevated expression of AXL was observed following BRAFi and MEKi (either single or combination) treatments in patient-derived xenografts (PDXs) [99][77]. To better understand the mechanisms behind resistance, Shaffer and colleagues compared the expression of resistance markers in pre-resistant cells and BRAFi-treated cells [92][50]. Interestingly, the number of resistance markers expressed increased from 72 in the pre-resistant cells to 600 (of the 1456 genes tested) in the post-drug-treatment cells by transcriptome analysis. Only 33 differentially accessible sites were discovered by comparing the non-resistant and pre-resistant cells versus more than 10,000 differentially accessible sites between pre- and post-drug-treatment using ATAC-seq, suggesting minute variations between the non-resistant and pre-resistant cells, and that the treatment induced phenotype switching resistance. Yet, whether the 33 differentially accessible sites between non-resistant and pre-resistant cells are sufficient to provoke a remarkable effect in drug resistance is arguable. In another study, resistance-conferring genetic alterations were evidenced in PDXs established from treatment-naïve patient samples, suggesting the existence of intrinsic resistant populations in melanoma patients [124][107]. Nevertheless, acquired resistance was observed in some PDXs too, indicating variabilities in causations of resistance to treatments. Meanwhile, other slow-cycling populations, such as JARID1Bhigh cells, CD133+, ABCB5+ and NGFR+ cells, have also been implicated in resistance to BRAFi and other anti-cancer drugs [49,76,78,125,126][49][83][85][108][109]. Furthermore, the PIK3CAE545K mutation was found to confer resistance to combined MEKi + CDK4i in a melanoma patient [127][110]. Nonetheless, aberrant expression of methyltransferases in melanomas could affect the sensitivity of melanoma cells to chemotherapy [128][111]. In addition, not only did pre-existing NGFRhigh melanoma cells show reduced sensitivity to the combination of BRAFi and MEKi but the cells were also resistant to antigen-specific cytotoxic CD8+ T cells [129][112]. Consistent with this finding, patients whose tumours are NGFRhigh were not responsive to anti-PD-1 and/or a combination of anti-PD-1 and anti-CTLA-4 therapy [129][112]. Furthermore, acquired JAK1 and JAK2 loss-of-function mutations were found in tumours after patients developed resistance to anti-PD-1 therapy [130][113]. The mutations were discovered by comparing the whole-exome sequencing data of tumour samples before treatment and after relapse. The cells with JAK1 mutations were resistant to interferon α, β and γ-induced growth arrest, while the cells with JAK2 mutations were insensitive to interferon γ-induced growth arrest. The same study also revealed the mutation of beta-2-microglobulin (B2M) in another patient’s baseline and matched relapse samples, where the loss of B2M reduced the immune-cell recognition of cancer cells. Nonetheless, there are ambiguous responses to immunotherapy due to tumour heterogeneity. For instance, data from a mouse study suggested that tumours with a low level of intratumoural heterogeneity may be more immunogenic and, hence, have an improved overall survival rate [105][102]. Lin and colleagues found that tumours with high heterogeneity were associated with decreased immune cell infiltration, immune response activation and overall patient survival [104][101]. In contrast, a positive correlation was observed between high mutational load and enhanced T cell infiltration in patient samples [35][100], indicating that tumours with a high mutational load may be more immunogenic. Despite the inconsistencies observed, it is possible that the identity of the genes mutated is potentially more pertinent in determining the treatment response regardless of the amount of mutational load [131][114]. Altogether, studies have shown that drug treatment could trigger secondary mutations in tumour cells, which lead to resistance. It is also evident that some tumour cells harbour pre-existing resistance, conferring genetic properties that reduce sensitivities to treatments and later lead to treatment resistance. The heterogeneous tumour environment similarly contributes to immunotherapy treatment resistance. The development of immunotherapy has greatly transformed the treatment for advanced melanoma and dramatically increased the overall survival rate for melanoma patients. However, there are still nearly 40% of advanced melanoma patients that do not respond initially to immunotherapies, with adverse side effects observed in 60% of the patients [132][115]. The reason why some patients respond well while others do not respond at all is still unclear, although it is clear that heterogeneity at the tumour and immune levels plays an important role.5. Conclusions and Implications
Heterogeneity within melanoma was initially described by Norris, an attentive general practitioner, in 1820 [133][116]. Since that time, the weresearchers have gained significantly more discerning information regarding melanoma development and tumour heterogeneity, particularly at the molecular level. However, many aspects of the development and role of heterogeneity remain unclear. For example, the true identity of each of the subpopulations within the patient tumour remains to be clarified. Further, the specific roles of communication between different subpopulations within the tumour and the various biological components within the tumour microenvironment (ECM and immune cells) are not well characterised. As more subpopulations develop in the tumour, the more challenges patients must face, such as tumour growth, metastasis, efficacy of the therapeutic treatments and, finally, resistance to treatment. These open questions must serve as a reminder that much more needs to be done in order to fill in the missing pieces and truly understand the role of heterogeneity in melanoma metastasis and treatment resistance in order to effectively tackle the disease. Effectively targeting the tumour heterogeneity in melanoma could lead to better therapeutic options and better outcomes for patients.References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Wood, J.M.; Jimbow, K.; Boissy, R.E.; Slominski, A.; Plonka, P.M.; Slawinski, J.; Wortsman, J.; Tosk, J. What’s the use of generating melanin? Exp. Dermatol. 1999, 8, 153–164.
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850.
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358.
- Weatherhead, S.C.; Haniffa, M.; Lawrence, C.M. Melanomas arising from naevi and de novo melanomas—Does origin matter? Br. J. Dermatol. 2007, 156, 72–76.
- Kiuru, M.; Tartar, D.M.; Qi, L.; Chen, D.; Yu, L.; Konia, T.; McPherson, J.D.; Murphy, W.J.; Fung, M.A. Improving classification of melanocytic nevi: Association of BRAF V600E expression with distinct histomorphologic features. J. Am. Acad. Dermatol. 2018, 79, 221–229.
- Bauer, J.; Curtin, J.; Pinkel, D.; Bastian, B.C. Congenital Melanocytic Nevi Frequently Harbor NRAS Mutations but no BRAF Mutations. J. Investig. Dermatol. 2007, 127, 179–182.
- Tschandl, P.; Berghoff, A.S.; Preusser, M.; Burgstaller-Muehlbacher, S.; Pehamberger, H.; Okamoto, I.; Kittler, H. NRAS and BRAF Mutations in Melanoma-Associated Nevi and Uninvolved Nevi. PLoS ONE 2013, 8, e69639.
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421.
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180.
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively Generated Damage to Cellular DNA by UVB and UVA Radiation. Photochem. Photobiol. 2015, 91, 140–155.
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310.
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; Soberino-García, J.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947.
- Osella-Abate, S.; Bertero, L.; Senetta, R.; Mariani, S.; Lisa, F.; Coppola, V.; Metovic, J.; Pasini, B.; Puig, S.S.; Fierro, M.T.; et al. TERT Promoter Mutations are Associated with Visceral Spreading in Melanoma of the Trunk. Cancers 2019, 11, 452.
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. New Engl. J. Med. 2015, 373, 1926–1936.
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263.
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218.
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113.
- Ding, L.; Kim, M.; Kanchi, K.L.; Dees, N.D.; Lu, C.; Griffith, M.; Fenstermacher, D.; Sung, H.; Miller, C.A.; Goetz, B.; et al. Clonal Architectures and Driver Mutations in Metastatic Melanomas. PLoS ONE 2014, 9, e111153.
- Janku, F. Tumor heterogeneity in the clinic: Is it a real problem? Ther. Adv. Med. Oncol. 2014, 6, 43–51.
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975.
- Ito, T.; Tanaka, Y.; Murata, M.; Kaku-Ito, Y.; Furue, K.; Furue, M. BRAF Heterogeneity in Melanoma. Curr. Treat. Options Oncol. 2021, 22, 20.
- Sakaizawa, K.; Goto, Y.; Kiniwa, Y.; Uchiyama, A.; Harada, K.; Shimada, S.; Saida, T.; Ferrone, S.; Takata, M.; Uhara, H.; et al. Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br. J. Cancer 2012, 106, 939–946.
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS Mutation Frequencies Among Primary Tumors and Metastases in Patients with Melanoma. J. Clin. Oncol. 2012, 30, 2522–2529.
- Yancovitz, M.; Litterman, A.; Yoon, J.; Ng, E.; Shapiro, R.L.; Berman, R.S.; Pavlick, A.C.; Darvishian, F.; Christos, P.; Mazumdar, M.; et al. Intra- and inter-tumor heterogeneity of BRAF(V600E)) mutations in primary and metastatic melanoma. PLoS ONE 2012, 7, e29336.
- Heinzerling, L.; Baiter, M.; Kühnapfel, S.; Schuler, G.; Keikavoussi, P.; Agaimy, A.; Kiesewetter, F.; Hartmann, A.; Schneider-Stock, R. Mutation landscape in melanoma patients clinical implications of heterogeneity of BRAF mutations. Br. J. Cancer 2013, 109, 2833–2841.
- Saint-Jean, M.; Quéreux, G.; Nguyen, J.M.; Peuvrel, L.; Brocard, A.; Vallée, A.; Knol, A.C.; Khammari, A.; Denis, M.G.; Dréno, B. Is a single BRAF wild-type test sufficient to exclude melanoma patients from vemurafenib therapy? J. Investig. Dermatol. 2014, 134, 1468–1470.
- Bradish, J.R.; Richey, J.D.; Post, K.M.; Meehan, K.; Sen, J.D.; Malek, A.J.; Katona, T.M.; Warren, S.; Logan, T.F.; Fecher, L.A.; et al. Discordancy in BRAF mutations among primary and metastatic melanoma lesions: Clinical implications for targeted therapy. Mod. Pathol. 2015, 28, 480–486.
- Yaman, B.; Kandiloglu, G.; Akalin, T. BRAF-V600 Mutation Heterogeneity in Primary and Metastatic Melanoma: A Study with Pyrosequencing and Immunohistochemistry. Am. J. Dermatopathol. 2016, 38, 113–120.
- Eriksson, H.; Zebary, A.; Vassilaki, I.; Omholt, K.; Ghaderi, M.; Hansson, J. BRAFV600E protein expression in primary cutaneous malignant melanomas and paired metastases. JAMA Dermatol. 2015, 151, 410–416.
- Chatterjee, A.; Stockwell, P.A.; Ahn, A.; Rodger, E.J.; Leichter, A.L.; Eccles, M.R. Genome-wide methylation sequencing of paired primary and metastatic cell lines identifies common DNA methylation changes and a role for EBF3 as a candidate epigenetic driver of melanoma metastasis. Oncotarget 2016, 8, 6085–6101.
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943.
- Sanborn, J.Z.; Chung, J.; Purdom, E.; Wang, N.J.; Kakavand, H.; Wilmott, J.S.; Butler, T.; Thompson, J.F.; Mann, G.J.; Haydu, L.E.; et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc. Natl. Acad. Sci. USA 2015, 112, 10995–11000.
- Rabbie, R.; Ansari-Pour, N.; Cast, O.; Lau, D.; Scott, F.; Welsh, S.J.; Parkinson, C.; Khoja, L.; Moore, L.; Tullett, M.; et al. Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nat. Commun. 2020, 11, 4306.
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. Bogdan, I.; Xin, H.; Burg, G.; Böni, R. Heterogeneity of allelic deletions within melanoma metastases. Melanoma Res. 2001, 11, 349–354.
- Bogdan, I.; Xin, H.; Burg, G.; Böni, R. Heterogeneity of allelic deletions within melanoma metastases. Melanoma Res. 2001, 11, 349–354. Harbst, K.; Lauss, M.; Cirenajwis, H.; Winter, C.; Howlin, J.; Törngren, T.; Kvist, A.; Nodin, B.; Olsson, E.; Häkkinen, J.; et al. Molecular and genetic diversity in the metastatic process of melanoma. J. Pathol. 2014, 233, 39–50.
- Harbst, K.; Lauss, M.; Cirenajwis, H.; Winter, C.; Howlin, J.; Törngren, T.; Kvist, A.; Nodin, B.; Olsson, E.; Häkkinen, J.; et al. Molecular and genetic diversity in the metastatic process of melanoma. J. Pathol. 2014, 233, 39–50. Lin, J.; Goto, Y.; Murata, H.; Sakaizawa, K.; Uchiyama, A.; Saida, T.; Takata, M. Polyclonality of BRAF mutations in primary melanoma and the selection of mutant alleles during progression. Br. J. Cancer 2011, 104, 464–468.
- Lin, J.; Goto, Y.; Murata, H.; Sakaizawa, K.; Uchiyama, A.; Saida, T.; Takata, M. Polyclonality of BRAF mutations in primary melanoma and the selection of mutant alleles during progression. Br. J. Cancer 2011, 104, 464–468. Manfredi, L.; Meyer, N.; Tournier, E.; Grand, D.; Uro-Coste, E.; Rochaix, P.; Brousset, P.; Lamant, L. Highly Concordant Results Between Immunohistochemistry and Molecular Testing of Mutated V600E BRAF in Primary and Metastatic Melanoma. Acta Derm. Venereol. 2016, 96, 630–634.
- Manfredi, L.; Meyer, N.; Tournier, E.; Grand, D.; Uro-Coste, E.; Rochaix, P.; Brousset, P.; Lamant, L. Highly Concordant Results Between Immunohistochemistry and Molecular Testing of Mutated V600E BRAF in Primary and Metastatic Melanoma. Acta Derm. Venereol. 2016, 96, 630–634. Busam, K.J.; Hedvat, C.; Pulitzer, M.; von Deimling, A.; Jungbluth, A.A. Immunohistochemical analysis of BRAF(V600E) expression of primary and metastatic melanoma and comparison with mutation status and melanocyte differentiation antigens of metastatic lesions. Am. J. Surg. Pathol. 2013, 37, 413–420.
- Busam, K.J.; Hedvat, C.; Pulitzer, M.; von Deimling, A.; Jungbluth, A.A. Immunohistochemical analysis of BRAF(V600E) expression of primary and metastatic melanoma and comparison with mutation status and melanocyte differentiation antigens of metastatic lesions. Am. J. Surg. Pathol. 2013, 37, 413–420. Gremel, G.; Lee, R.J.; Girotti, M.R.; Mandal, A.K.; Valpione, S.; Garner, G.; Ayub, M.; Wood, S.; Rothwell, D.; Fusi, A.; et al. Distinct subclonal tumour responses to therapy revealed by circulating cell-free DNA. Ann. Oncol. 2016, 27, 1959–1965.
- Gremel, G.; Lee, R.J.; Girotti, M.R.; Mandal, A.K.; Valpione, S.; Garner, G.; Ayub, M.; Wood, S.; Rothwell, D.; Fusi, A.; et al. Distinct subclonal tumour responses to therapy revealed by circulating cell-free DNA. Ann. Oncol. 2016, 27, 1959–1965. Sensi, M.; Nicolini, G.; Petti, C.; Bersani, I.; Lozupone, F.; Molla, A.; Vegetti, C.; Nonaka, D.; Mortarini, R.; Parmiani, G.; et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 2006, 25, 3357–3364.
- Sensi, M.; Nicolini, G.; Petti, C.; Bersani, I.; Lozupone, F.; Molla, A.; Vegetti, C.; Nonaka, D.; Mortarini, R.; Parmiani, G.; et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 2006, 25, 3357–3364. Wilmott, J.S.; Tembe, V.; Howle, J.R.; Sharma, R.; Thompson, J.F.; Rizos, H.; Lo, R.S.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Intratumoral Molecular Heterogeneity in a BRAF-Mutant, BRAF Inhibitor-Resistant Melanoma: A Case Illustrating the Challenges for Personalized Medicine. Mol. Cancer Ther. 2012, 11, 2704–2708.
- Wilmott, J.S.; Tembe, V.; Howle, J.R.; Sharma, R.; Thompson, J.F.; Rizos, H.; Lo, R.S.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Intratumoral Molecular Heterogeneity in a BRAF-Mutant, BRAF Inhibitor-Resistant Melanoma: A Case Illustrating the Challenges for Personalized Medicine. Mol. Cancer Ther. 2012, 11, 2704–2708. Mitra, A.; Andrews, M.C.; Roh, W.; De Macedo, M.P.; Hudgens, C.W.; Carapeto, F.; Singh, S.; Reuben, A.; Wang, F.; Mao, X.; et al. Spatially resolved analyses link genomic and immune diversity and reveal unfavorable neutrophil activation in melanoma. Nat. Commun. 2020, 11, 1839.
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048.
- Rastetter, M.; Schagdarsurengin, U.; Lahtz, C.; Fiedler, E.; Marsch, W.C.; Dammann, R.; Helmbold, P. Frequent intra-tumoural heterogeneity of promoter hypermethylation in malignant melanoma. Histol. Histopathol. 2007, 22, 1005–1015.
- Sigalotti, L.; Fratta, E.; Coral, S.; Tanzarella, S.; Danielli, R.; Colizzi, F.; Fonsatti, E.; Traversari, C.; Altomonte, M.; Maio, M. Intratumor Heterogeneity of Cancer/Testis Antigens Expression in Human Cutaneous Melanoma Is Methylation-Regulated and Functionally Reverted by 5-Aza-2′-deoxycytidine. Cancer Res. 2004, 64, 9167–9171.
- Pan, M.; Reid, M.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101.
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell 2010, 141, 583–594.
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825.
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435.
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598.
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic Heterogeneity among Tumorigenic Melanoma Cells from Patients that Is Reversible and Not Hierarchically Organized. Cancer Cell 2010, 18, 510–523.
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656.
- Eichhoff, O.M.; Zipser, M.C.; Xu, M.; Weeraratna, A.T.; Mihic, D.; Dummer, R.; Hoek, K.S. The immunohistochemistry of invasive and proliferative phenotype switching in melanoma: A case report. Melanoma Res. 2010, 20, 349–955.
- Widmer, D.S.; Cheng, P.F.; Eichhoff, O.M.; Belloni, B.C.; Zipser, M.C.; Schlegel, N.C.; Javelaud, D.; Mauviel, A.; Dummer, R.; Hoek, K.S. Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment. Cell Melanoma Res. 2012, 25, 343–353.
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683.
- Chapman, A.; del Ama, L.F.; Ferguson, J.; Kamarashev, J.; Wellbrock, C.; Hurlstone, A. Heterogeneous Tumor Subpopulations Cooperate to Drive Invasion. Cell Rep. 2014, 8, 688–695.
- Campbell, N.R.; Rao, A.; Hunter, M.V.; Sznurkowska, M.K.; Briker, L.; Zhang, M.; Baron, M.; Heilmann, S.; Deforet, M.; Kenny, C.; et al. Cooperation between melanoma cell states promotes metastasis through heterotypic cluster formation. Dev. Cell 2021, 56, 2808–2825.e10.
- Salti, G.I.; Manougian, T.; Farolan, M.; Shilkaitis, A.; Majumdar, D.; Das Gupta, T.K. Micropthalmia transcription factor: A new prognostic marker in intermediate-thickness cutaneous malignant melanoma. Cancer Res. 2000, 60, 5012–5016.
- Ennen, M.; Keime, C.; Gambi, G.; Kieny, A.; Coassolo, S.; Thibault-Carpentier, C.; Margerin-Schaller, F.; Davidson, G.; Vagne, C.; Lipsker, D.; et al. MITF-High and MITF-Low Cells and a Novel Subpopulation Expressing Genes of Both Cell States Contribute to Intra- and Intertumoral Heterogeneity of Primary Melanoma. Clin. Cancer Res. 2017, 23, 7097–7107.
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., II; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196.
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; LaRue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439.
- Goding, C.R. A picture of Mitf in melanoma immortality. Oncogene 2011, 30, 2304–2306.
- Rowling, E.J.; Miskolczi, Z.; Nagaraju, R.; Wilcock, D.; Wang, P.; Telfer, B.; Li, Y.; Lasheras-Otero, I.; Redondo-Muñoz, M.; Sharrocks, A.D.; et al. Cooperative behaviour and phenotype plasticity evolve during melanoma progression. Pigment Cell Melanoma Res. 2020, 33, 695–708.
- Lister, J.A.; Capper, A.; Zeng, Z.; Mathers, M.E.; Richardson, J.; Paranthaman, K.; Jackson, I.J.; Patton, E.E. A Conditional Zebrafish MITF Mutation Reveals MITF Levels Are Critical for Melanoma Promotion vs. Regression In Vivo. J. Investig. Dermatol. 2014, 134, 133–140.
- Travnickova, J.; Wojciechowska, S.; Khamseh, A.; Gautier, P.; Brown, D.V.; Lefevre, T.; Brombin, A.; Ewing, A.; Capper, A.; Spitzer, M.; et al. Zebrafish MITF-Low Melanoma Subtype Models Reveal Transcriptional Subclusters and MITF-Independent Residual Disease. Cancer Res. 2019, 79, 5769–5784.
- Pinner, S.; Jordan, P.; Sharrock, K.; Bazley, L.; Collinson, L.; Marais, R.; Bonvin, E.; Goding, C.; Sahai, E. Intravital Imaging Reveals Transient Changes in Pigment Production and Brn2 Expression during Metastatic Melanoma Dissemination. Cancer Res. 2009, 69, 7969–7977.
- Goodall, J.; Carreira, S.; Denat, L.; Kobi, D.; Davidson, I.; Nuciforo, P.; Sturm, R.A.; Larue, L.; Goding, C.R. Brn-2 Represses Microphthalmia-Associated Transcription Factor Expression and Marks a Distinct Subpopulation of Microphthalmia-Associated Transcription Factor–Negative Melanoma Cells. Cancer Res. 2008, 68, 7788–7794.
- Thurber, A.E.; Douglas, G.; Sturm, E.C.; Zabierowski, S.E.; Smit, D.J.; Ramakrishnan, S.N.; Hacker, E.E.; Leonard, J.H.; Herlyn, M.; Sturm, R.A. Inverse expression states of the BRN2 and MITF transcription factors in melanoma spheres and tumour xenografts regulate the NOTCH pathway. Oncogene 2011, 30, 3036–3048.
- Fane, M.; Chhabra, Y.; Hollingsworth, D.E.; Simmons, J.; Spoerri, L.; Oh, T.G.; Chauhan, J.; Chin, T.; Harris, L.; Harvey, T.J.; et al. NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF. eBioMedicine 2017, 16, 63–75.
- Boyle, G.M.; Woods, S.L.; Bonazzi, V.F.; Stark, M.S.; Hacker, E.; Aoude, L.G.; Dutton-Regester, K.; Cook, A.L.; Sturm, R.A.; Hayward, N.K. Melanoma cell invasiveness is regulated by miR-211 suppression of the BRN2 transcription factor. Pigment Cell Melanoma Res. 2011, 24, 525–537.
- Golan, T.; Messer, A.R.; Amitai-Lange, A.; Melamed, Z.; Ohana, R.; Bell, R.E.; Kapitansky, O.; Lerman, G.; Greenberger, S.; Khaled, M.; et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis via Notch Signaling Inhibition of MITF. Mol. Cell 2015, 59, 664–676.
- Smith, M.P.; Rana, S.; Ferguson, J.; Rowling, E.J.; Flaherty, K.T.; Wargo, J.A.; Marais, R.; Wellbrock, C. A PAX3/BRN2 rheostat controls the dynamics of BRAF mediated MITF regulation in MITF(high)/AXL(low) melanoma. Pigment Cell Melanoma Res. 2019, 32, 280–291.
- Simmons, J.; Pierce, C.J.; Al-Ejeh, F.; Boyle, G.M. MITF and BRN2 contribute to metastatic growth after dissemination of melanoma. Sci. Rep. 2017, 7, 1–12.
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A Melanoma Cell State Distinction Influences Sensitivity to MAPK Pathway Inhibitors. Cancer Discov. 2014, 4, 816–827.
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712.
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.-Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212.
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480.
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349.
- Wang, S.; Tang, L.; Lin, J.; Shen, Z.; Yao, Y.; Wang, W.; Tao, S.; Gu, C.; Ma, J.; Xie, Y.; et al. ABCB5 promotes melanoma metastasis through enhancing NF-kappaB p65 protein stability. Biochem. Biophys. Res. Commun. 2017, 492.
- Wilson, B.J.; Saab, K.R.; Ma, J.; Schatton, T.; Pütz, P.; Zhan, Q.; Murphy, G.F.; Gasser, M.; Waaga-Gasser, A.M.; Frank, N.Y.; et al. ABCB5 Maintains Melanoma-Initiating Cells through a Proinflammatory Cytokine Signaling Circuit. Cancer Res. 2014, 74, 4196–4207.
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946.
- Jamal, S.M.E.; Alamodi, A.; Wahl, R.U.; Grada, Z.; Shareef, M.A.; Hassan, S.Y.; Murad, F.; Hassan, S.L.; Santourlidis, S.; Gomez, C.R.; et al. Melanoma stem cell maintenance and chemo-resistance are mediated by CD133 signal to PI3K-dependent pathways. Oncogene 2020, 39, 5468–5478.
- Zimmerer, R.M.; Korn, P.; Demougin, P.; Kampmann, A.; Kokemüller, H.; Eckardt, A.M.; Gellrich, N.-C.; Tavassol, F. Functional features of cancer stem cells in melanoma cell lines. Cancer Cell Int. 2013, 13, 78.
- Simbulan-Rosenthal, C.M.; Dougherty, R.; Vakili, S.; Ferraro, A.M.; Kuo, L.-W.; Alobaidi, R.; Aljehane, L.; Gaur, A.; Sykora, P.; Glasgow, E.; et al. CRISPR-Cas9 Knockdown and Induced Expression of CD133 Reveal Essential Roles in Melanoma Invasion and Metastasis. Cancers 2019, 11, 1490.
- Lai, C.-Y.; Schwartz, B.E.; Hsu, M.-Y. CD133+ Melanoma Subpopulations Contribute to Perivascular Niche Morphogenesis and Tumorigenicity through Vasculogenic Mimicry. Cancer Res. 2012, 72, 5111–5118.
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nat. Cell Biol. 2010, 466, 133–137.
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109.
- Su, Y.; Bintz, M.; Yang, Y.; Robert, L.; Ng, A.H.C.; Liu, V.; Ribas, A.; Heath, J.R.; Wei, W. Phenotypic heterogeneity and evolution of melanoma cells associated with targeted therapy resistance. PLoS Comput. Biol. 2019, 15, e1007034.
- Perego, M.; Maurer, M.; Wang, J.X.; Shaffer, S.; Müller, A.C.; Parapatics, K.; Li, L.; Hristova, D.; Shin, S.; Keeney, F.; et al. A slow-cycling subpopulation of melanoma cells with highly invasive properties. Oncogene 2018, 37, 302–312.
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A Isozymes are Markers of Human Melanoma Stem Cells and Potential Therapeutic Targets. Stem Cells 2012, 30, 2100–2113.
- Wang, Y.; Leonard, M.K.; Snyder, D.E.; Fisher, M.L.; Eckert, R.L.; Kaetzel, D.M. NME1 Drives Expansion of Melanoma Cells with Enhanced Tumor Growth and Metastatic Properties. Mol. Cancer Res. 2019, 17, 1665–1674.
- Snyder, D.; Wang, Y.; Kaetzel, D.M. A rare subpopulation of melanoma cells with low expression of metastasis suppressor NME1 is highly metastatic in vivo. Sci. Rep. 2020, 10, 1971.
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278.
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301.
- Luo, C.; Lim, J.-H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.; Puigserver, P. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426.
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120.
- Bernsen, M.R.; Diepstra, J.H.S.; van Mil, P.; Punt, C.J.; Figdor, C.G.; van Muijen, G.N.; Adema, G.J.; Ruiter, D.J. Presence and localization of T-cell subsets in relation to melanocyte differentiation antigen expression and tumour regression as assessed by immunohistochemistry and molecular analysis of microdissected T cells. J. Pathol. 2003, 202, 70–79.
- Bosisio, F.M.; Antoranz, A.; Van Herck, Y.; Bolognesi, M.M.; Marcelis, L.; Chinello, C.; Wouters, J.; Magni, F.; Alexopoulos, L.G.; Stas, M.; et al. Functional heterogeneity of lymphocytic patterns in primary melanoma dissected through single-cell multiplexing. eLife 2020, 9, e53008.
- Mitra, A.; Andrews, M.C.; Roh, W.; De Macedo, M.P.; Hudgens, C.W.; Carapeto, F.; Singh, S.; Reuben, A.; Wang, F.; Mao, X.; et al. Spatially resolved analyses link genomic and immune diversity and reveal unfavorable neutrophil activation in melanoma. Nat. Commun. 2020, 11, 1839. Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 10.
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 10. Lin, Z.; Meng, X.; Wen, J.; Corral, J.M.; Andreev, D.; Kachler, K.; Schett, G.; Chen, X.; Bozec, A. Intratumor Heterogeneity Correlates with Reduced Immune Activity and Worse Survival in Melanoma Patients. Front. Oncol. 2020, 10, 596493.
- Lin, Z.; Meng, X.; Wen, J.; Corral, J.M.; Andreev, D.; Kachler, K.; Schett, G.; Chen, X.; Bozec, A. Intratumor Heterogeneity Correlates with Reduced Immune Activity and Worse Survival in Melanoma Patients. Front. Oncol. 2020, 10, 596493. Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e21.
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e21. Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107.
- Sekulic, A.; Hingorani, P.; Lenkiewicz, E.; Holley, T.; Barrett, M.; Zismann, V.; Trent, J. Clonal evolution underlying trans-placental transferand vemurafenib resistance in melanoma. Society for Melanoma Research 2012 Congress. Pigment Cell Melanoma Res. 2012, 25, 836–903.
- Khaliq, M.; Manikkam, M.; Martinez, E.D.; Fallahi-Sichani, M. Epigenetic modulation reveals differentiation state specificity of oncogene addiction. Nat. Commun. 2021, 12, 1536.
- Marin-Bejar, O.; Rogiers, A.; Dewaele, M.; Femel, J.; Karras, P.; Pozniak, J.; Bervoets, G.; Van Raemdonck, N.; Pedri, D.; Swings, T.; et al. Evolutionary predictability of genetic versus nongenetic resistance to anticancer drugs in melanoma. Cancer Cell 2021, 39, 1135–1149.e8.
- El-Khattouti, A.; Sheehan, N.T.; Monico, J.; Drummond, H.A.; Haikel, Y.; Brodell, R.T.; Megahed, M.; Hassan, M. CD133 + melanoma subpopulation acquired resistance to caffeic acid phenethyl ester-induced apoptosis is attributed to the elevated expression of ABCB5: Significance for melanoma treatment. Cancer Lett. 2015, 357, 83–104.
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4448–4459.
- Romano, G.; Chen, P.L.; Song, P.; McQuade, J.L.; Liang, R.J.; Liu, M.; Roh, W.; Duose, D.Y.; Carapeto, F.C.L.; Li, J.; et al. A Preexisting Rare PIK3CA(E545K) Subpopulation Confers Clinical Resistance to MEK plus CDK4/6 Inhibition in NRAS Melanoma and Is Dependent on S6K1 Signaling. Cancer Discov. 2018, 8, 556–567.
- Campagna, R.; Salvolini, E.; Pompei, V.; Pozzi, V.; Salvucci, A.; Molinelli, E.; Brisigotti, V.; Sartini, D.; Campanati, A.; Offidani, A.; et al. Nicotinamide N-methyltransferase gene silencing enhances chemosensitivity of melanoma cell lines. Pigment Cell Melanoma Res. 2021, 34, 1039–1048.
- Boshuizen, J.; Vredevoogd, D.W.; Krijgsman, O.; Ligtenberg, M.A.; Blankenstein, S.; De Bruijn, B.; Frederick, D.T.; Kenski, J.C.N.; Parren, M.; Brüggemann, M.; et al. Reversal of pre-existing NGFR-driven tumor and immune therapy resistance. Nat. Commun. 2020, 11, 3946.
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829.
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241.
- Asher, N.; Ben-Betzalel, G.; Lev-Ari, S.; Shapira-Frommer, R.; Steinberg-Silman, Y.; Gochman, N.; Schachter, J.; Meirson, T.; Markel, G. Real World Outcomes of Ipilimumab and Nivolumab in Patients with Metastatic Melanoma. Cancers 2020, 12, 2329.
- Rebecca, V.W.; Sondak, V.K.; Smalley, K.S. A brief history of melanoma: From mummies to mutations. Melanoma Res. 2012, 22, 114–122.