Alpha-synuclein (α-syn) is a small protein composed of 140 amino acids and belongs to the group of intrinsically disordered proteins. It is a soluble protein that is highly expressed in neurons and expressed at low levels in glial cells. The monomeric protein aggregation process induces the formation of oligomeric intermediates and proceeds towards fibrillar species. These α-syn conformational species have been detected in the extracellular space and mediate consequences on surrounding neurons and glial cells. In particular, higher-ordered α-syn aggregates are involved in microglial and oligodendrocyte activation, as well as in the induction of astrogliosis. These phenomena lead to mitochondrial dysfunction, reactive oxygen and nitrogen species formation, and the induction of an inflammatory response, associated with neuronal cell death. Several receptors participate in cell activation and/or in the uptake of α-syn, which can vary depending on the α-syn aggregated state and cell types. The receptors involved in this process are of outstanding relevance because they may constitute potential therapeutic targets for the treatment of PD and related synucleinopathies.
- extracellular alpha-synuclein
- oligomers
- fibrils
- astrocytes
- oligodendrocytes
- microglia
1. Introduction
1.1. Synucleinopathies
1.2. Alpha-Synuclein and Aggregation Process
α-Syn is a small protein composed of 140 amino acids (14 KDa) and is abundant in many regions of the brain [16][17][16,17]. α-Syn is an intrinsically disordered protein, with no defined structure. The protein has three well-characterized regions: the N-terminal region (1–60 amino acids), the hydrophobic region or non-A beta component of the Alzheimer’s disease amyloid (61–95 amino acids), and the C-terminal region (96–140 amino acids) [18][19][20][21][18,19,20,21]. The N-terminal region includes six copies of the repeat KTKEGV and is the fragment of the protein where familial mutations of the α-syn gene related to PD have been identified [22]. The hydrophobic region is the amyloidogenic part of the protein, related to the ability of α-syn to form fibrils in vitro and in vivo. This region is what distinguishes α-syn from the other members of the synuclein family [22][23][24][25][26][22,23,24,25,26]. The C-terminal region is rich in proline residues and has a high content of acidic amino acids, such as glutamic and aspartic acid. This contributes to the extremely low isoelectric point of α-syn (pI: 4.7) [27]. α-Syn fibril formation follows a nucleation-dependent pathway that involves many prefibrillar intermediates. The fibrillation of monomeric α-syn requires the formation of a discrete number of soluble oligomeric intermediates [28]. Oligomeric prefibrillar species include a group of intermediates of variable size and morphology [28][29][30][31][32][33][34][35][36][28,29,30,31,32,33,34,35,36]. Lansbury´s group has proposed that spheroidal α-syn soluble oligomers are rich in β-sheet structure and that the conversion from monomer to oligomer involves a secondary structural transition from the natively unfolded protein to predominantly β-sheets [37]. Oligomers of α-syn are formed from approximately 30 to 35 monomers and have a molecular weight of 440,000 Da. α-Syn fibrils are much larger, with about 8300 monomers per fibril and a molecular weight of around 120,000,000 Da [38]. Many reports suggest that these oligomeric species are responsible for α-syn toxicity [37][39][40][41][37,39,40,41]. It has been demonstrated that α-syn soluble oligomers disrupt membranes [37][41][37,41] and cause cell death both in vitro [30][31][30,31] and in animal models [39][40][39,40]. Therefore, processes that increase α-syn oligomer concentration, stabilize its conformation, or decrease its clearance will probably induce toxicity [35]. However, the exact role of oligomeric species in α-syn pathology is still unclear. Nevertheless, the idea that α-syn soluble oligomers are the proximal toxic species has been questioned since it has been shown that fibrils of α-syn can also induce toxicity, promote the seeding of endogenous α-syn, and may have prion-like effects [14][15][32][33][42][14,15,32,33,42].2. Extracellular α-Synuclein
2.1. Putative Mechanisms of α-Syn Uptake in Cells of the Nervous System
α-Syn is a cytosolic protein that is poorly expressed in astrocytes, microglia, and oligodendrocytes [43][44][43,44]. However, it is abundantly expressed in neurons in the central nervous system (CNS). Although α-syn has no known signaling sequence, α-syn can be released from neuronal cells in small amounts via unconventional exocytosis under normal physiological conditions [45]. In pathological conditions, α-syn monomers and aggregates may be released in larger quantities and endocytosed by neighboring cells, leading to the formation of LB-like inclusions [46]. Both forms of the protein, monomeric and higher-order aggregated species, have been found in the lumen of vesicles [45]. Recent evidence showed that α-syn can propagate through neurons in the central nervous system. First, LBs were found in grafted neurons in PD patients treated with embryonic cell transplants [47][48][47,48]. Second, animal studies showed that brain inoculation of fibrillar α-syn led to the propagation of α-syn to anatomically interconnected areas of the brain, and in humans, there is evidence of trans-synaptically spreading of α-syn pathology [34][49][50][51][52][34,49,50,51,52]. Third, significant amounts of α-syn soluble oligomers have been detected in the plasma and cerebrospinal fluid (CSF) of patients with PD [53].2.2. Glial Cell Uptake of Extracellular α-Syn and Activation
2.2.1. Role of Astrocytes
Astrocytes outnumber neurons in the CNS and are responsible for a wide variety of important functions, including regulation of blood flow, maintenance of the blood–brain barrier (BBB), and maintenance of the composition of the extracellular environment of ions [54][65]. Recent studies suggest that astrocytes play important roles in modulating neurotransmission, cell signaling, inflammation, synapse modulation, and metabolite and electrolyte homeostasis. Damage to the CNS due to injury or disease may result in molecular, cellular, and functional changes in astrocytes, leading to ”reactive astrogliosis”. The process of astrocyte activation can be divided into three main stages or features: (i) morphological changes and cytokine production, (ii) cell proliferation, and (iii) cell migration. Some characteristics that describe reactive astrogliosis are: astrocyte hypertrophy, development of processes and cell proliferation, increased expression of the cytoskeleton glial fibrillary acidic protein (GFAP), and alterations in gene expression [54][55][56][57][65,66,68,69]. As well as other glial cells, astrocytes do not express α-syn or express it at very low levels [58][70]. However, the uptake of wild-type or mutant α-syn by astrocytes induces astrocyte reactivity, exhibiting neurotoxicity or inducing inflammation [59][60][71,72]. In the development of synucleinopathies, astrocytes may be activated, either by α-syn or by activated microglia [61][62][67,73]. Different α-syn aggregated forms activate glial cells to induce an inflammatory response [58][59][70,71]. Astrocytes exposed to neuron-derived α-syn aggregates underwent changes in their gene expression profiles with the induction of different proinflammatory cytokines and chemokines [63][74]. Reactive astrocytes can promote the release of proinflammatory cytokines and induce the production of reactive oxygen species, which will in turn affect neuronal survival and neuronal functions [59][63][71,74]. Oxidative stress has been implicated in the pathogenic mechanisms of PD and many other neurodegenerative diseases [64][65][75,76]. In response to oxidative stress, the levels of numerous cytoprotective products are increased via alteration of the Keap1 and Nrf2 system [66][77]. The formation of peroxynitrite and radicals derived from its homolysis leads to the oxidation and nitration of proteins [67][68][69][78,79,80]. In particular, for α-syn, the exposure of the protein to nitrating agents in vitro results in cross-linking and the formation of high-molecular-mass α-syn aggregates [70][81]. Pathological α-syn accumulation impairs the redox homeostasis in the nervous system; an increase in nuclear localization of NRF2 in post-mortem PD midbrain was detected [64][75]. The relevance of astrocytes in this scenario is also their participation in the clearance of neuronal α-syn, revealing an important role of astrocytes in the regulation of neuronal α-syn [71][82]. A relationship between mitochondrial dysfunction and α-syn has been previously reported in PD. However, most mitochondrial studies in PD were performed in neuronal cells. PD patients present an accumulation of α-syn in mitochondria and decreased complex I activity, while mice overexpressing mutated A53T α-syn have reduced complex IV activity [72][73][84,85]. Astrocytes’ cytoarchitecture dramatically changes upon exposure to oligomeric and fibrillar α-syn, with the generation of flat and polyhedral cells, retraction of the soma and nuclei, and formation of long thin processes. There is an increase in the immunostaining of the GFAP protein in astrocytes upon oligomer and fibrillar α-syn exposure along with the morphological changes. α-Syn soluble oligomers and fibrils induce the mRNA of TNF-alpha and IL-1β at similar levels to the ones obtained with LPS on astrocytes. All α-syn conformers induced the formation of reactive oxygen and nitrogen species, but only the soluble oligomeric forms led to mitochondrial dysfunction in cortical astrocytes.2.2.2. Role of Oligodendrocytes
Oligodendrocytes are glial cells that are responsible for the myelination of axons in the CNS, having an important role in their development, maintenance, and regeneration. Oligodendrocytes undergo a complex process of proliferation, migration, and differentiation that leads to their mature form. They also provide trophic support to neurons by releasing lactate [74][75][76][77][78][88,89,90,91,92]. The connection between α-syn and oligodendrocytes comes from pathology [79][80][93,94]. MSA is a progressive and severe neurodegenerative disorder that is clinically characterized by variable degrees of parkinsonism, cerebellar ataxia, and dysautonomia [81][95]. The hallmark of the disease is the presence of glial cytoplasmic inclusions (GCIs), which are intracellular protein aggregates, mainly composed of α-syn, located in oligodendrocytes [7][82][7,96]. Further components of GCIs are ubiquitin and other proteins, such as leucine-rich repeat serine/threonine-protein LRRK2, heat shock proteins, microtubule-associated protein tau, and prion disease-linked 14-3-3 protein, among others [82][96]. Analysis of single-nucleotide polymorphisms (SNPs) in the SNCA gene, the gene that encodes for α-syn, has identified an association between certain α-syn SNPs and an increased risk for the development of MSA [83][97]. Even though α-syn mRNAs and protein were detected in rat brain oligodendrocytes [84][98], α-syn expression was not detected in oligodendrocytes from healthy and MSA human brains [43]. This implies that endogenous α-syn is not enough for the formation of intracellular aggregates associated with pathology. In response to cellular stress, oligodendrocytes suffer from oligodendroglial dysfunction. iIn a transgenic mouse model expressing α-syn in oligodendrocytes (under the control of the MBP promoter), there was a decrease in the expression of neurotrophic factors, especially glial-derived neurotrophic factor (GDNF) released from oligodendrocytes, providing new insight into the possible pathogenic mechanisms of oligodendroglial α-synucleinopathies [85][101].2.2.3. Role of Microglia
Microglia are phagocytic cells of the brain that regulate brain development, the maintenance of neuronal networks, and injury repair. Accumulation and activation of microglia in the CNS have been termed microgliosis. Activated microglia change the movement of their processes from undirected to targeted towards the injured site [86][106]. Microglial cells express a wide range of immune receptors, such as pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) or tissue damage–associated molecular patterns (DAMPs). Microglia PRRs include toll-like receptors (TLRs), particularly TLR4 and TLR1/2 and their co-receptors [87][107]. The relationship between α-syn, microglia, and disease arises from the observation that PD patients demonstrate a marked increase in activated microglia with increased expression and concentration of proinflammatory cytokines [88][89][90][104,108,109]. In addition, reactive microglia assemble close to LBs in PD patients [91][110]. Moreover, α-syn leads to microglial activation in mouse models of protein overexpression prior to dopaminergic neuronal death [92][111]. Microglia exposed to α-syn soluble oligomers upregulate the expression of genes encoding TLR and the proinflammatory cytokines TNF-α and IL-1β [93][112]. They also present morphological changes indicative of microglial activation. Microglial activation is also associated with the generation of reactive oxygen and nitrogen species [94][113]. In the substantia nigra pars compacta of MSA mice, increased expression of inducible nitric oxide synthase was detected [95][114]. The activation of microglia and the proinflammatory response produced can accelerate the loss of dopaminergic neurons and the progression of synucleinopathies [96][105]. The different conformers of α-syn, mainly soluble oligomers, induce a specific response. In contrast, monomeric α-syn does not induce detectable microglial activation but promotes microglial phagocytosis [97][115]. α-Syn preformed fibrils (PFFs) also induced the activation of microglia [98][116]. Information from proteomics indicates that α-syn PFF leads to expression changes of microglial genes involved in RNA binding, mitochondrial stress, and lysosomal and autophagic functions, shedding light on the pathways involved in α-syn PFF activation of microglia [99][117].2. Receptors for Extracellular α-Syn
Table 1 and Figure 12 summarize the extracellular receptors discussed here, identifying the α-syn conformer and the cell type involved. The LAG3 receptor belongs to the immunoglobulin superfamily. It is highly expressed in some immune organs, including the spleen and the thymus, and also in the central nervous system [100][101][119,120]. LAG3 can be expressed on neuronal cells and in microglia [102][121]. This receptor regulates T cell immune responses and immune homeostasis, mainly by inhibiting T cell activation and proliferation. LAG3 demonstrated the highest ratio of selectivity for α-syn PFF over monomeric α-syn. The internalization of α-syn PFF in neurons involves LAG3, since the deletion of LAG3 reduces the endocytosis of α-syn PFF, and this is specific for α-syn PFF. Neuron-to-neuron transmission of α-syn and the induction of neurotoxicity are attenuated by the deletion of LAG3 [103][63]. The lack of LAG3 delayed the α-syn PFF-induced loss of dopamine neurons, as well as biochemical and behavioral deficits in vivo [103][63]. Some scholarsauthors described an impairment in the pole test of animals injected with PFF, which was also prevented by LAG3 deletion [103][63].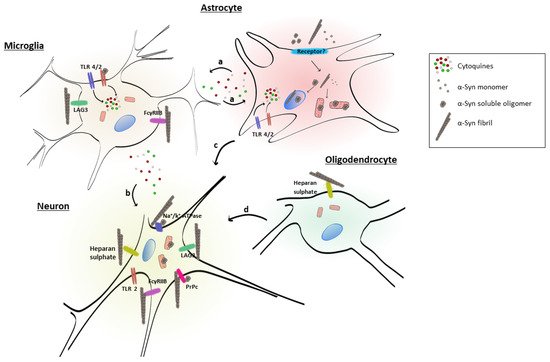
| Receptor | α-Syn Conformer | Cell Type | Reference | |||||
|---|---|---|---|---|---|---|---|---|
| Lymphocyte-activation gene 3 (LAG3) | Fibrils | Neuron, microglia | [102][103] | [63,121] | ||||
| Cellular prion protein (PrPc) | Fibrils, soluble oligomers | Neurons | [104][105] | [122,123] | ||||
| Heparan sulfate | Fibrils | Neuron, oligodendrocytes | [106] | [64] | ||||
| Toll-like receptor 4 (TLR4) | n.d. | Microglia | [107] | [61] | ||||
| Toll-like receptor 2 (TLR2) | Soluble oligomers | Microglia | [108] | [118] | ||||
| a3-subunit of Na | + | /K | + | -ATPase | Fibrils, soluble oligomers | Neurons | [109] | [124] |
| FcƔRIIB | Fibrils | Microglia, neurons | [110] | [125] |
These data suggest that extracellular α-syn fibrils can bind to LAG3 and contribute to protein-induced dopaminergic neuronal loss and neurotoxicity [103][63]. This receptor may play a role in α-syn spreading pathology and neurodegeneration in PD and could be considered as a therapeutic target to avoid α-syn pathology.