PolyADP-ribosylation is an evolutionary conserved, reversible post-translational modification of proteins. Numerous nuclear proteins act as substrates of the abundant nuclear polyADP-ribose polymerase 1 (PARP1). In this modification, negatively charged ADP-ribose chains constructed on chromatin-bound proteins, cause their repulsion from the negatively charged DNA. In accordance, polyADP-ribosylation is a post-translational modification of proteins that causes relaxation of the highly condensed structure of the chromatin. Histone H1, which is bound to the linker DNA, located between the nucleosomes, is a prominent substrate of PARP1. Recent findings identified eviction of polyADP-ribosylated H1 from the linker DNA in response to a variety of stimulations, resulting in DNA relaxation.
- PARP1 activation
- H1 polyADP-ribosylation
- signal transduction pathways
1. Introduction
2. PARP1 Activation by Ca2+ Released from Intracellular IP3-Gated Stores
In a variety of cell types treated with hormones, growth factors, neurotransmitters, and membrane depolarization in excitable cells, G-protein-coupled receptors and receptor tyrosine kinases are activated [19][20]. Their activation initiates the activation of a variety of second messengers in networks of signal transduction pathways involving protein kinases and phosphatases [19][20]. In one of these signaling mechanisms, the multi-targeted phospholipase-C cleaves phosphatidyl inositols that are embedded in the cell membrane, including phosphatidylinositol 4,5-bisphosphate (PIP2), which is cleaved into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) [19][20][21]. DAG remains embedded in the membrane and acts as an anchoring molecule, and IP3 is released into the cytoplasm [19][20][21]. The binding of IP3 to specific receptors in Ca2+ gated stores in the endoplasmic reticulum induces intracellular Ca2+ release [21][22][23][24]. IP3-induced Ca2+ release from its stores in the endoplasmic reticulum, which includes the perinuclear membrane, activates a variety of Ca2+-dependent enzymes, including kinases and phosphatases participating in a variety of signal transduction pathways [22][23][24][25]. PARP1 was activated in response to Ca2+ release from the perinuclear stores into the nucleoplasm [25][26]. Ca-induced PARP1 polyADP-ribosylation was measured in isolated nuclei of a variety of cell types [25][26]. The effect of Ca2+ on PARP1 polyADP-ribosylation resembled the well-documented co-factor activity of Mg2+ ions in in-vitro polyADP-ribosylation of PARP1 [26]. Divalent cations frequently act as co-factors in enzymatic reactions [27]. The IP3-induced release of Ca2+ into the nucleoplasm enhanced the polyADP-ribosylation of PARP1 in nuclei of rat cerebral neurons, as well as in nuclei of neuronal cells of the marine slug Aplysia [25] (Figure 1).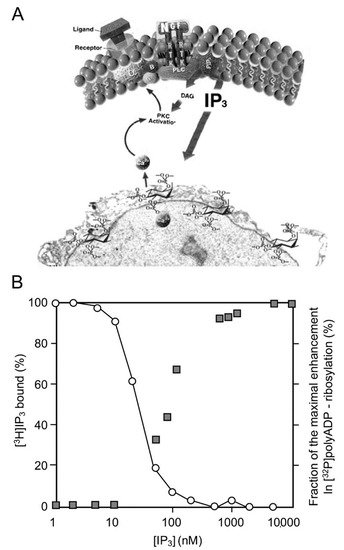
3. A Long-Lasting PARP1 Activation by Phosphorylated Erk
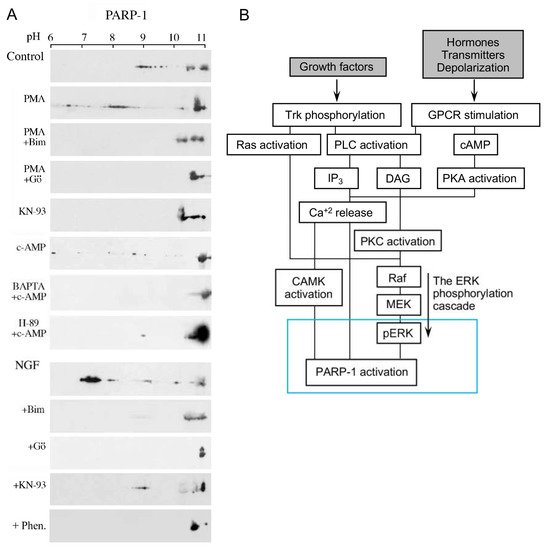
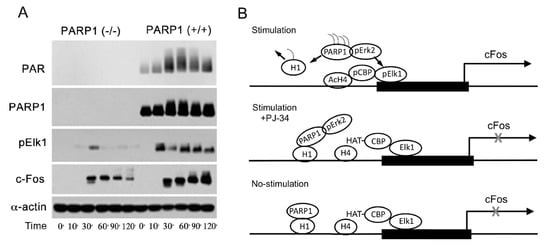
Signals inducing phosphorylation of MEK dimers, which are bound to dimers of Erk (extracellular signal-regulated kinase) in the cytoplasm, release the phosphorylated Erk [32][33][34][35]. The released phosphorylated Erk, which lacks the nuclear localization signal (NLS), is translocated into the nucleus [33][34][35][36]. Phosphorylated Erk has a long-lasting activity in the nucleus, despite lacking the NLS signal [35][36]. A variety of transcription factors promoting the expression of numerous genes are activated by Erk-induced phosphorylation [35][37], including those promoting immediate-early genes (IEG) expression [37][38]. Phosphorylated Erk1/2 are apparently translocated into the nucleus as homodimers [33][34] by specific transporters [33][34][35][36]. In view of the similarities between the protein binding sites of Erk1 and Erk2, as well as the major role of Erk2 activity in mammals [39], the effect of phosphorylated Erk2 on PARP1 activity has been explored. Co-immunoprecipitation of phosphorylated Erk2 with PARP1 or polyADP-ribosylated PARP1 was measured in a variety of cell types [10][15][40]. Furthermore, consensus docking sites of phosphorylated Erk were identified in PARP1 [10]. They include four sites that partially match the known docking motifs of phosphorylated Erk, found in its substrates 633KYPKK637, 683KK684, 747KKPPLL752, and 1007FNF. They are located in the WGR domain, helical domain (HD), and catalytic (CAT) domain of PARP1 (aa556–1014) [10][41][42][43][44]. In addition, a negatively charged protein-binding domain in Erk (CRS/CD region) was involved in its binding to the docking sites in PARP1 [10][15] (Figure 2). Phosphorylated Erk was co-immunoprecipitated with PARP1 in nuclear extracts of a variety of stimulated cell types, and MEK inhibitors interfered with their binding [10][15][16]. Furthermore, in a cell-free system, recombinant PARP1 was activated and highly polyADP-ribosylated upon binding of recombinant phosphorylated Erk2 [10][15]. In accordance, PARP1 activation measured in a variety of stimulated cell types was dependent on the phosphorylation of MEK [10][14][15][16][40] (Figure 2C). In addition, a long-lasting activity of phosphorylated Erk in the nuclei of these cells was dependent on PARP1 [10][15][16][17][18]. A variety of transcription factors, promoters, and modulators in the chromatin, which are prominent substrates of phosphorylated Erk, were hardly phosphorylated in PARP1 KO cells [10][18], indicating the PARP1-dependent activity of phosphorylated Erk in the chromatin. PolyADP-ribosylated PARP1 apparently acts as a platform for phosphorylated Erk, enabling phosphorylation of Erk substrates while causing local chromatin relaxation.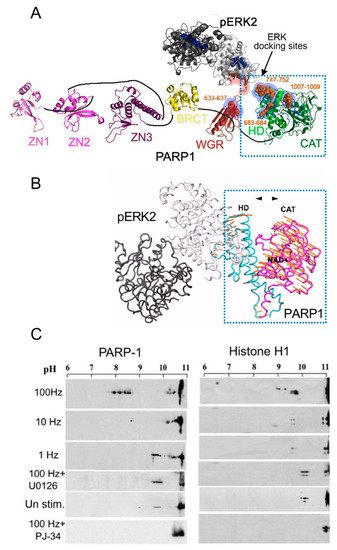
References
- Langelier, M.-F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes:regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198.
- Kallberg, T.; Langelier, M.-F.; Pascal, J.; Schuler, H. Structural biology of writers, readers, and erasers in mono- and poly(ADP-ribose)mediated signaling. Mol. Asp. Med. 2013, 34, 1089–1108.
- Dantzer, F.; Amé, J.C.; Schreiber, V.; Nakamura, J.; Ménissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 2006, 409, 493–510.
- Luscher, B.; Butepage, M.; Eckei, L.; Kreig, S.; Verheugd, P. ADP ribosylation, a multifaceted posttranslational modification involved in the control of cell physiology in health and disease. Chem. Rev. 2018, 118, 1092–1136.
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732.
- Poirier, G.G.; deMurcia, G.; Jongstra-Bilen, J.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427.
- Tulin, A.; Spradling, A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puf loci. Science 2003, 299, 560–562.
- Kraus, W.L.; Lis, J.T. PARP goes transcription. Cell 2003, 113, 677–683.
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal binding of PARP-1 and histone H1 at promoters specifes transcriptional outcomes. Science 2008, 319, 819–821.
- Visochek, L.; Grigoryan, G.; Kalal, A.; Milshtein-Parush, H.; Gazit, N.; Slutsky, I.; Yeheskel, A.; Shainberg, A.; Castiel, A.; Seger, R. A PARP1-ERK2 synergism is required for the induction of LTP L. Sci. Rep. (Nat.) 2016, 6, 24950.
- Izzo, A.; Schneider, R. The role of linker histone H1modifications in the regulation of gene expression and chromatin dynamics. Biochim. Biophys. Acta 2016, 1859, 486–495.
- Azad, G.K.; Ito, K.; Sailaja, T.; Biran, A.; Nissim-Rafinia, M.; Yamada, Y.; Brown, D.T.; Takizawa, T.; Meshorer, E. PARP1-dependent eviction of the linker histone H1mediates immediate early gene expression during neuronal activation. J. Cell. Biol. 2017, 217, 473–481.
- Fontan-Lozano, A.; Suarez-Pereira, I.; Horillo, A.; del-Pozo-Martin, Y.; Hmadch, A. Histone H1 Poly-Ribosylation Regulates the Chromatin Alterations Required for Learning Consolidation. J. Neurosci. 2010, 30, 13305–13313.
- Cohen-Armon, M.; Yeheskel, A.; Pascal, J.M. Signal-induced PARP1-Erk synergism mediates IEG expression. Signal Transduct. Target. Ther. 2019, 4, 8.
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-Independent PARP-1 Activation by Phosphorylated ERK2 Increases Elk1 Activity: A Link to Histone Acetylation. Mol. Cell 2007, 25, 297–308.
- Visochek, l.; Steingart, R.; Vulin-Shultzman, I.; Klein, R.; Priel, E.; Gozes, I.; Cohen-Armon, M. PolyADP-ribosylation is involved in neurotrophic activity. J. Neurosci. 2005, 25, 7420–7428.
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560.
- Visochek, L.; Cohen-Armon, M. PARP1-Erk synergism in proliferating cells. Oncotarget 2018, 9, 29140–29145.
- Berridge, M.J.; Irvine, R.F. Inositol phosphates and cell signaling. Nature 1989, 341, 197–204.
- Banno, Y.; Nagao, S.; Katada, T.; Nagata, K.; Ui, M.; Nozawa, Y. Stimulation by GTP-binding proteins (Gi, Go) of partially purified phospholipase C activity from human platelet membranes. Biochem. Biophys. Res. Commun. 1987, 146, 861–869.
- Exton, J.H. Signaling through phosphatidylcholine breakdown. J. Biol. Chem. 1990, 265, 1–4.
- Hennager, D.J.; Welsh, M.J.; Delise, S. Changes in either cytosolic or nucleoplasmic inositol 1,4,5-triphosphate levels can control nuclear Ca+2 concentration. J. Biol. Chem. 1995, 270, 4959–4962.
- Malviya, A.N.; Rogue, P.J. “Tell me where is calcium bred”: Clarifying the roles of nuclear calcium. Cell 1998, 92, 17–23.
- Gerasimenko, O.V.; Gerasimenko, V.J.; Tepikin, A.V.; Petersen, O.H. ATP-dependent accumulation and inositol trisphos phateor cyclic ADPribose-mediated release of Ca+2 from the nuclear envelope. Cell 1995, 80, 439–444.
- Cohen-Armon, M. PARP1 activation is required for long-term memory. In Long-Term Memory: Mechanisms, Types and Disorders. Series: Neurosci. Res. Progress, Perspectives on Cognitive Psychology; Alexandrov, A.K., Fedoseev, L.M., Eds.; NOVA Sci. Pub.: New York, NY, USA, 2012.
- Homburg, S.; Visochek, L.; Moran, N.; Dantzer, F.; Priel, E.; Asculai, E.; Schwartz, D.; Rotter, V.; Dekel, N.; Cohen-Armon, M. A Fast Signal–Induced Activation of Poly(Adp-Ribose) Polymerase: A Novel Downstream Target of Phospholipase C. J. Cell Biol. 2000, 150, 293–308.
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thoronton, J.M. Metal ions in biological catalysis: From Enzyme databases to general principles. J. Biol. Inorg. Chem. 2008, 13, 1205–12018.
- Ju, B.-G.; Schum, D.; Song, E.J.; Lee, K.-J.; Ross, D.W.; Glass, C.K.; Rosenfeld, M.G. Activating the PARP1-sensor component of the Groucho/TLE1 core pressor complex mediates a CaMKinase dependent neurogenic gene activation pathway. Cell 2004, 119, 815–829.
- Payne, M.E.; Fong, Y.L.; Ono, T.; Collbran, R.J.; Kemp, B.E.; Shoderling, T.R.; Means, A.R. Calcium/calmodulin-dependent protein kinase II. J. Biol Chem. 1988, 263, 7190–7199.
- Kuo, P. The mechanism of protein kinase C activation. Trends Neurosci. 1989, 12, 425–432.
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18.
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase:Multiple substrates regulate cellular functions. Growth Factors 2006, 24, 21–44.
- Khokhlachev, A.V.; Canagarajah, B.; Wilsbacher, J.; Robinson, M.; Atkinson, M.; Goldsmith, E.; Cobb, M. Phosphorylation of MAP kinase Erk2 promotes its homodimerization and nuclear translocation. Cell 1998, 91, 605–615.
- Casar, B.; Pinto, A.; Crespo, P. ERK dimers and Scaffold proteins, Un-expected partners for a forgotten (cytoplasmic) task. Cell Cycle 2009, 8, 7–17.
- Bhalla, U.S.; Iyengar, R. Emergent properties of networks of biological signaling pathways. Science 1999, 283, 381–387.
- Sasagawa, S.; Ozaki, Y.-I.; Fujita, K.; Kuroda, S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol. 2005, 7, 365–373.
- Buchwalter, G.; Gross, C.; Wasylyk, B. Ets ternary complex transcription factors. Gene 2004, 324, 1–14.
- Pouyssegur, J.; Lenormand, P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signaling. Eur. J. Biochem. 2003, 270, 3291–3299.
- Busca, R.; Pouyssegur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific roles or functional redundancy. Front. Cell Dev. Biol. 2016, 4, 1–23.
- Geistrikh, I.; Visochek, L.; Klein, R.; Miller, L.; Mittelman, L.; Shainberg, A.; Cohen-Armon, M. Ca+2-induced PARP-1 activation and ANF expression are coupled events in cardiomyocytes. Biochem. J. 2011, 438, 337–347.
- Tanoue, T.; Adachi, M.; Moriguchi, T.; Nishida, E. A conserved docking motif in MAP kinases common to substrates activetors and regulators. Nat. Cell Biol. 2000, 2, 110–116.
- Jacobs, D.; Glossip, D.; Xing, H.; Muslin, A.J.; Kornfeld, K. Multiple docking sites on substrate proteins form modular system that mediates recognition by Erk MAP kinase. Gene Dev. 1999, 13, 163–175.
- Tanoue, T.; Nishida, E. Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol. Ther. 2002, 2–3, 193–202.
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O. Anisotropy of fluctuation dynamics of protein with elastic network model. Biophys. J. 2001, 80, 505–515.