Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Tarek Bismar and Version 2 by Amina Yu.
The ETS-related gene (ERG) is proto-oncogene that is classified as a member of the ETS transcription factor family, which has been found to be consistently overexpressed in about half of the patients with clinically significant prostate cancer (PCa). The overexpression of ERG can mostly be attributed to the fusion of the ERG and transmembrane serine protease 2 (TMPRSS2) genes, and this fusion is represented about 85% of all gene fusions observed in prostate cancer. Clinically, individuals with ERG gene fusion are mostly documented to have advanced tumor stages, increased mortality, and higher rates of metastasis in non-surgical cohorts.
- gene fusion
- prostate cancer
- tumorigenesis
- ()
1. Structural Characteristics and Allosteric Autoinhibition of ERG
The ERG transcription factor has a primary structure that consists of 486 amino acids and a corresponding molecular weight of 54kDa [1][21]. A distinguishing characteristic of ETS family proteins is the presence of a DNA-binding domain called the ETS DNA binding domain (EBD). This EBD domain is made of 85 amino acids and contains 3 alpha-helices that are further supported by a 4-stranded anti-parallel beta sheet [2][24]. The EBD domain plays a critical role in DNA recognition as well as in AP-1 and co-activator recruitment [3][25]. Within the EBD, there are three highly conserved tryptophan residues that serve as a hydrophobic core to facilitate the helix–turn–helix binding domain in proteins [4][26]. ERG analysis by means peptide sequencing has also predicted the presence of phosphorylation sites for protein kinase C and a pointed (PNT) domain in the N-terminus [1][21]. The PNT domains are a part of a larger sterile alpha motif (SAM) family that is involved in many diverse protein–protein interactions that can allow for self-association [5][27]. This PNT domain has also been shown to facilitate the heterodimerization of ERG with other proteins, including other members of the ETS family, DNA-dependent kinases, AP-1 complex, and the androgen receptor (AR) [6][28].
2. The Function of ETS-Related Gene (ERG) in Normal Cell Types
ERG has a multitude of physiological functions that differ based on the type of cells or the organism’s developmental stage. During embryogenesis, ERG has been observed to be highly expressed in the mesoderm and endothelium, where it plays a crucial role in vasculogenesis and in the development of bone [7][29]. In adults, it regulates vascular homeostasis and angiogenesis by activating the transcription of endothelial specific genes, such as vascular endothelial (VE)-cadherin, an adhesion molecule that promotes vascular stability by maintaining and controlling endothelial cell contact [8][30]. In addition to this, VE-cadherin also plays a central role in cell proliferation and apoptosis and modulates endothelial growth factor receptor functions [9][31]. Other endothelial genes that are positively regulated by ERG include the vascular endothelial growth factor (VEGF), von Willebrand factor, and endoglin, which are all involved in endothelial cell differentiation and angiogenesis [4][10][26,32].
3. Prominent TMPRSS2-ERG Gene Fusion Found in PCa
In prostate cancer cells, a surprisingly common occurrence involves the fusion of ERG to TMPRSS2, which forms the fusion product of TMPRSS2-ERG. The most common mechanism by which these two genes fuse involves the deletion of intronic sequences on the long arm of chromosome 21 via an intron deletion between TMPRSS2 and ERG on chromosome 21q22.2-3 (Figure 1). This fusion mechanism has been identified as being prevalent in approximately 50% of prostate cancer patients [11][33]. The frequent occurrence of this fusion protein can be attributed to the presence of a homogenous deletion site that is present between ERG and TMPRSS2 [12][34]. Moreover, this deletion site is separated into two different classifications according to various start sites. In both of the deletion products, the 5′ end of the TMPRSS2 gene has been ligated to the 3′ end of ERG. TMPRSS2-ERG fusion results in ERG overexpression due to the androgen responsive promoter of the TMPSS2 gene allowing for the constitutive transcription of ERG, which has been shown to be correlated with increased cell proliferation, cell invasion, angiogenesis, and invasiveness in PCa cells [13][14][35,36]. In addition, this TMPRSS2-ERG fusion enhances the transcription and activates downstream oncogenes [15][37].
4. Functional ERG Overexpression in Prostate Cancer Cells
In prostate cancer cells, ERG overexpression increases the rate of epithelial to mesenchymal transitions via the EMT pathway, enhancing the ability of PCa cells to invade and metastasize. ERG achieves this by upregulating matrix metalloproteinases (MMPs), CXCR4, and Osteopontin (OPN), which have been correlated with higher rates of cell invasion and metastasis among patients [16][38]. Additionally, the TMPRSS2-ERG pathway reveals the epithelial to mesenchymal transition via the ZEB1/ZEB2 axis in PCa [17][39]. The constitutive expression of ERG also hyperactivates the inflammatory pathway in PCa cells by binding to Toll-like receptor 4; this activates the NF-kb pathway, increasing the transcription of target genes such as TNFA, IL6, BCLXL, BCL2, BCLXS, XIAP, and VEGF [18][19][40,41]. These proteins trigger tumor growth and progression by enhancing cell proliferation, survival, and angiogenesis. Tumor growth is further accentuated by the activation of the EZH2 promoter by ERG. This relieves the epigenetic inhibition of tumor suppressor genes such as NKX3.1, resulting in the constitutive expression of the TMPRSS2-ERG fusion gene. In addition to its role in regulating tumor cell invasion and proliferation, ERG also plays an important role in negatively regulating tumor cell differentiation by inhibiting the transcription of genes such as KLK3/PSA and SLC45A3/Prostein [20][42]. Altogether, the overexpression of ERG in prostate cancer is a key modulator of tumor progression and aggressiveness, as it can regulate the transcription of the proteins that mediate inflammation, cell invasion, differentiation, and oncogenesis [21][43].
5. TMPRSS2-ERG Fusion Upregulates CXCR4 Enhances Tumor Adhesion and Aggregation
Previous onstudies have been concluded that there are eight ERG/Ets factor binding sites near the promoter of chemokine receptor type 4 (CXCR4) (Figure 13) and that TMPRSS2-ERG expression enhances the function and expression of CXCR4 [22][23][93,94]. CXCL12 is a known CXCR4 receptor ligand, and the interactions between the two functions enhance tumor aggressiveness and increase the ability of cancer cells to adhere to the extracellular matrix [24][25][95,96]. The CXCR4/CXCL12 axis has been shown to increase MMP expression, which promotes cell migration and growth in PCa [22][93]. An analysis of PCa demographics revealed relatively higher CXCR4 expression during PCa progression and in metastatic bone tissue compared to benign PCa cells that were reported. In vitro, a direct correlation between TMPRSS-ERG and CXCR4 was observed when ERG was knocked down [25][96].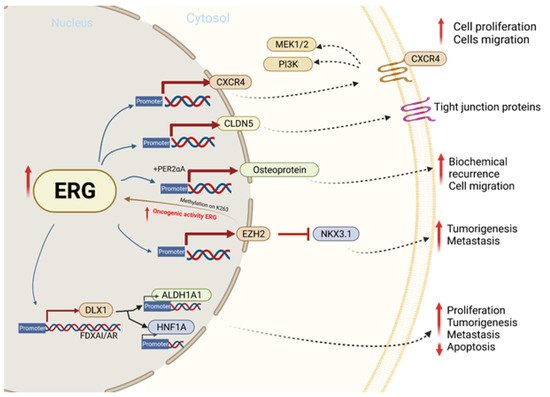
Figure 13. The molecular mechanism involved in ERG overexpression and related outcomes in tumor cell proliferation, migration, invasion, and metastasis.