Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Shin Takasawa.
Calcium (Ca2+) is a ubiquitous and fundamental signaling component that is utilized by cells to regulate a diverse range of cellular functions, such as insulin secretion from pancreatic β-cells of the islets of Langerhans. Cyclic ADP-ribose (cADPR), synthesized from NAD+ by ADP-ribosyl cyclase family proteins, such as the mammalian cluster of differentiation 38 (CD38), is important for intracellular Ca2+ mobilization for cell functioning. cADPR induces Ca2+ release from endoplasmic reticulum via the ryanodine receptor intracellular Ca2+ channel complex, in which the FK506-binding protein 12.6 works as a cADPR-binding regulatory protein.
- CD38
- cADPR
- RYR2
- FKBP12.6
- pancreatic β cells
- CD157
1. Introduction
Changes in cytosolic Ca
2+
are indispensable for normal cell function, such as secretion, proliferation, neuronal guidance, cell death, and development. Typically, cells respond to a Ca
2+
signal that is generated inside the cell in response to the activation of a wide variety of extracellular signals, including those involved in nutrient, neurotransmitter, hormonal, and sensory signaling. In most cases, the initial Ca
2+
signal generated in the cell is a specific increase in cytoplasmic Ca
2+
concentrations resulting from the release of Ca
2+
from internal Ca
2+
stores (mainly the endoplasmic reticulum) or the entry of Ca
2+
from the external spaces across the plasma membrane. Both routes involve the movement of Ca
2+
through Ca
2+
channels that are localized within these cellular membranes. While intracellular Ca
2+
release from the endoplasmic reticulum occurs via channels activated by inositol 1,4,5-trisphosphate (IP3), cyclic ADP-ribose (cADPR), or Ca
2+
itself (Ca
2+
-induced Ca
2+
release), Ca
2+
influx across the plasma membrane is achieved via numerous types of Ca
2+
channels, including voltage-gated Ca
2+
channels and store-operated Ca
2+
channels as well as a variety of ligand-gated cation channels.
2. cADPR as an Intracellular Ca
2. cADPR as an Intracellular Ca
2+
-Releasing Messenger
Changes in cytosolic Ca2+
levels govern a multitude of cellular processes. Many extracellular stimuli mediate complex changes in Ca
2+
2+
from the intracellular Ca
2+
store. In addition to IP3, cADPR and nicotinic acid adenine dinucleotide phosphate (NAADP) play crucial roles in the generation of agonist-evoked Ca
2+
signals. cADPR induces Ca
2+ signaling and have been implicated in a variety of processes ranging from bacterial clearance to social behavior [5]. ADP-ribosyl cyclase (EC 3.2.2.6) activity was originally identified in sea urchin egg homogenates [4], but an enzyme with the activity was later purified [6,7] and cloned [8,9] from Aplysia ovotestes. The enzyme was shown to catalyze the cyclization of NAD
signaling and have been implicated in a variety of processes ranging from bacterial clearance to social behavior [5]. ADP-ribosyl cyclase (EC 3.2.2.6) activity was originally identified in sea urchin egg homogenates [4], but an enzyme with the activity was later purified [6][7] and cloned [8][9] from Aplysia ovotestes. The enzyme was shown to catalyze the cyclization of NAD
+ to form cADPR and the replacement of the nicotinamide moiety in NADP with nicotinic acid to form NAADP. Based on amino acid sequence similarity, it became evident that the mammalian protein cluster of differentiation 38 (CD38; ADP-ribosyl cyclase 1; EC 3.2.2.6) [10,11,12,13] and cluster of differentiation 157 (CD157; ADP-ribosyl cyclase 2; EC 3.2.2.6) [14,15] were also ADP-ribosyl cyclase in mammalian cells.
3. cADPR in Insulin Secretion from Pancreatic β-Cells
Glucose induces an increase in intracellular Ca
to form cADPR and the replacement of the nicotinamide moiety in NADP with nicotinic acid to form NAADP. Based on amino acid sequence similarity, it became evident that the mammalian protein cluster of differentiation 38 (CD38; ADP-ribosyl cyclase 1; EC 3.2.2.6) [10][11][12][13] and cluster of differentiation 157 (CD157; ADP-ribosyl cyclase 2; EC 3.2.2.6) [14][15] were also ADP-ribosyl cyclase in mammalian cells.
3. cADPR in Insulin Secretion from Pancreatic β-Cells
Glucose induces an increase in intracellular Ca2+
concentration in pancreatic islet β-cells and induces the secretion of insulin. Ashcroft et al. first explained the importance of the increase in the Ca
2+
concentration in 1984 [16]. The millimolar concentration of ATP is produced by the process of glucose metabolism in pancreatic β-cells. As a result, there is the closure of ATP-sensitive potassium (KATP) channels, which results in membrane depolarization, the influx of Ca
2+
through voltage-dependent Ca
2+
channels, and an increase in the cytosolic Ca
2+
concentration that triggers the exocytosis of insulin granules. In 1993, another model of insulin secretion by glucose via cADPR-mediated Ca
2+
mobilization from an intracellular Ca
Figure 1
. According to this model, ATP inhibits the cADPR hydrolase of CD38, causing the accumulation of cADPR, which acts as a second messenger for Ca
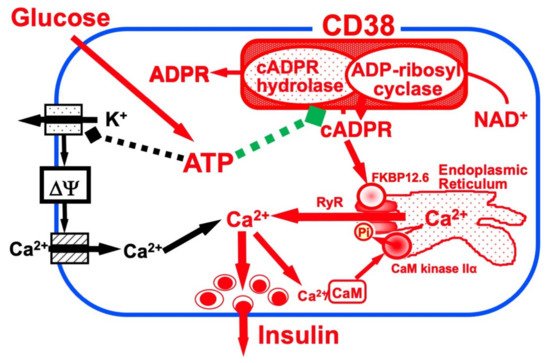
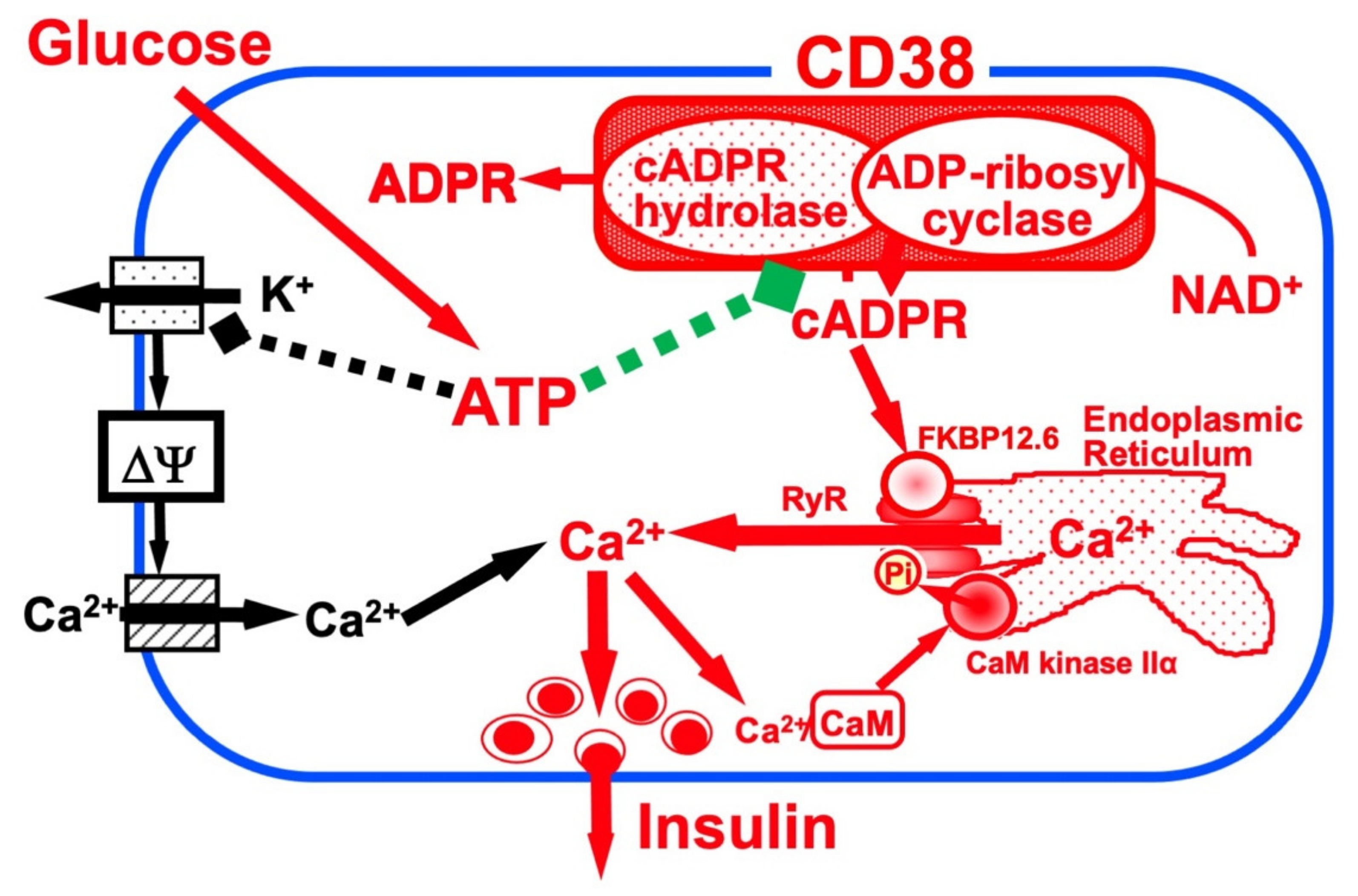
Figure 1. Insulin secretion by glucose stimulation in pancreatic β-cells (adapted from [33]). The insulin secretion via the CD38-cADPR signal system is shown in red. cADPR, produced from NAD
Insulin secretion by glucose stimulation in pancreatic β-cells [33]. The insulin secretion via the CD38-cADPR signal system is shown in red. cADPR, produced from NAD
+
by the ADP-ribosyl cyclase of CD38, binds to FKBP12.6 to release Ca
2+
, dissociating FKBP12.6 from RyR [25]. CaM kinase II phosphorylates RyR to sensitize and activate the Ca
2+
2+
, released from intracellular stores and/or supplied from extracellular sources, further activates CaM kinase II and amplifies the process. In this way, Ca
2+
-induced Ca
2+
release (CICR) can be explained in glucose-induced insulin secretion in pancreatic β-cells. The conventional insulin secretion mechanism by Ca
2+
influx from extracellular sources [16] is shown in black.
The first important issue is whether the accumulation of cADPR is actually caused by glucose stimulation in pancreatic islets. In the laboratory, the researchers assayed the cADPR content in islets isolated from normal rats (Wistar) and mice (C56BL/6J) that were incubated with low (2.8 mM) and high (20 mM) glucose by radioimmunoassay using an anti-cADPR antibody. The cADPR content in the high glucose-treated islets was rapidly increased within 5 min. In contrast, the cADPR content in the low glucose-treated islets was not [28].
Using the rat islet microsome Ca
2+
releasing system, fluo 3 (2,2′-{[2-(2-{2-[Bis(caboxymethyl)amino]-5-}2,7-dichloro-6-hydroxy-3-oxo-3H-xanthen-9-yl)phenoxy]ethoxy}-4-methylphenyl]azanediyl}diacetic acid) fluorescence showed cADPR released Ca
2+
, and after the addition of IP3, the islet microsomes were still responsive to cADPR. In contrast, IP3 caused a release of Ca
2+
from cerebellum microsomes, and cADPR also caused a release of Ca
2+
. Heparin, an inhibitor of IP3 binding to its receptor, blocked the IP3-induced Ca
2+
release from cerebellum microsomes but did not block the cADPR-induced Ca
2+
release. These results indicate that islet microsomes respond to cADPR but not to IP3. In contrast, cerebellum microsomes respond to both cADPR and IP3, but cADPR induces the Ca
2+
release via a different mechanism than that utilized by IP3.
The effect of cADPR on insulin secretion was examined using digitonin-permeabilized rat pancreatic islets. cADPR as well as Ca
2+
induced insulin secretion, but IP3 did not. The combined addition of cADPR and Ca
2+
did not induce significantly more insulin secretion than the addition of cADPR or Ca
2+
alone. The cADPR-induced insulin secretion was inhibited by the addition of EGTA. These results indicated cADPR induced Ca
2+
release from islet microsomes and the increment of Ca
2+
resulted in insulin secretion from pancreatic islets [17].
From these results, glucose-induced insulin secretion via cADPR formation from NAD
+
and cADPR-induced Ca
2+
mobilization from the endoplasmic reticulum was proposed. Concerning the second messenger role of cADPR for glucose-induced insulin secretion, some researchers reported that cADPR-induced intracellular Ca
2+ release was not observed in ob/ob mouse islets and rat insulinoma-derived RINm5F cells [19,35,36,37,38]. Malaisse et al. [39,40] measured the cADPR content in rat islets and reported that it appeared not to be significantly affected by glucose concentrations. On the other hand, the fasting of the rats before the isolation of islets and the usage of Hanks’ solution containing 2.8 mM glucose during the islet isolation may account for the rapid and significant increase in cADPR content in the islets in response to glucose stimulation. Furthermore the researchers determined the cADPR content by assessing the recovery of cADPR in the extraction and concentration procedures, and they did not. Differences in the experimental conditions may be responsible for the different results [19,28]. Nevertheless, the reports that Balb/c mouse islets showed increases in glucose-induced cADPR production, the intracellular concentration of Ca
release was not observed in ob/ob mouse islets and rat insulinoma-derived RINm5F cells [19][35][36][37][38]. Malaisse et al. [39][40] measured the cADPR content in rat islets and reported that it appeared not to be significantly affected by glucose concentrations. On the other hand, the fasting of the rats before the isolation of islets and the usage of Hanks’ solution containing 2.8 mM glucose during the islet isolation may account for the rapid and significant increase in cADPR content in the islets in response to glucose stimulation. Furthermore the researchers determined the cADPR content by assessing the recovery of cADPR in the extraction and concentration procedures, and they did not. Differences in the experimental conditions may be responsible for the different results [19][28]. Nevertheless, the reports that Balb/c mouse islets showed increases in glucose-induced cADPR production, the intracellular concentration of Ca
2+
, and insulin secretion [41] and that human insulin promoter-SV40 large T transgene-introduced C57BL/6 mouse pancreatic β-cell-derived MIN6 cells, which show glucose-induced insulin secretion, showed a dramatic Ca
2+ mobilization in response to cADPR via the ryanodine receptor (RyR) [42,43] despite the lack of response to IP3 [43]. These results indicate that the CD38-cADPR signal system is functioning in glucose-induced insulin secretion in pancreatic β-cells.
4. CD38 as a Major Enzyme for the Synthesis of cADPR
CD38 is a 300-amino-acid protein and was first recognized as a human leukocyte surface antigen. CD38 was found to express in a variety of tissues and cells, including pancreatic β-cells [12,20,44]. The researchers and others have found that CD38 has both ADP-ribosyl cyclase for cADPR synthesis from NAD
mobilization in response to cADPR via the ryanodine receptor (RyR) [42][43] despite the lack of response to IP3 [43]. These results indicate that the CD38-cADPR signal system is functioning in glucose-induced insulin secretion in pancreatic β-cells.
4. CD38 as a Major Enzyme for the Synthesis of cADPR
CD38 is a 300-amino-acid protein and was first recognized as a human leukocyte surface antigen. CD38 was found to express in a variety of tissues and cells, including pancreatic β-cells [12][20][44]. The researchers and others have found that CD38 has both ADP-ribosyl cyclase for cADPR synthesis from NAD+ and cADPR hydrolase for the hydrolysis of cADPR to form ADP-ribose [11,12,13,21,45]. Using purified human CD38 protein, it was found that millimolar concentrations of ATP inhibit the cADPR hydrolase activity of CD38, competing with the substrate, cADPR [12,26]. The competitive inhibition of the cADPR hydrolysis by ATP suggests that ATP and cADPR bind to the same site of CD38. The researchers purified human recombinant CD38, incubated with an ATP analogue, 5′-p-fluorosulfonylbenzoyladenosine, and identified the binding site for ATP and/or cADPR as the lysine-129 of CD38 [26]. From these results and other available evidence, the researchers proposed that CD38 catalyzes the formation of cADPR from NAD
and cADPR hydrolase for the hydrolysis of cADPR to form ADP-ribose [11][12][13][21][45]. Using purified human CD38 protein, it was found that millimolar concentrations of ATP inhibit the cADPR hydrolase activity of CD38, competing with the substrate, cADPR [12][26]. The competitive inhibition of the cADPR hydrolysis by ATP suggests that ATP and cADPR bind to the same site of CD38. The researchers purified human recombinant CD38, incubated with an ATP analogue, 5′-p-fluorosulfonylbenzoyladenosine, and identified the binding site for ATP and/or cADPR as the lysine-129 of CD38 [26]. From these results and other available evidence, the researchers proposed that CD38 catalyzes the formation of cADPR from NAD
+
and also the hydrolysis of the cADPR to ADP-ribose. As shown in
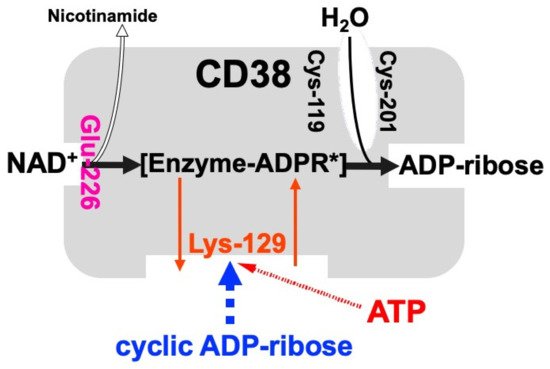
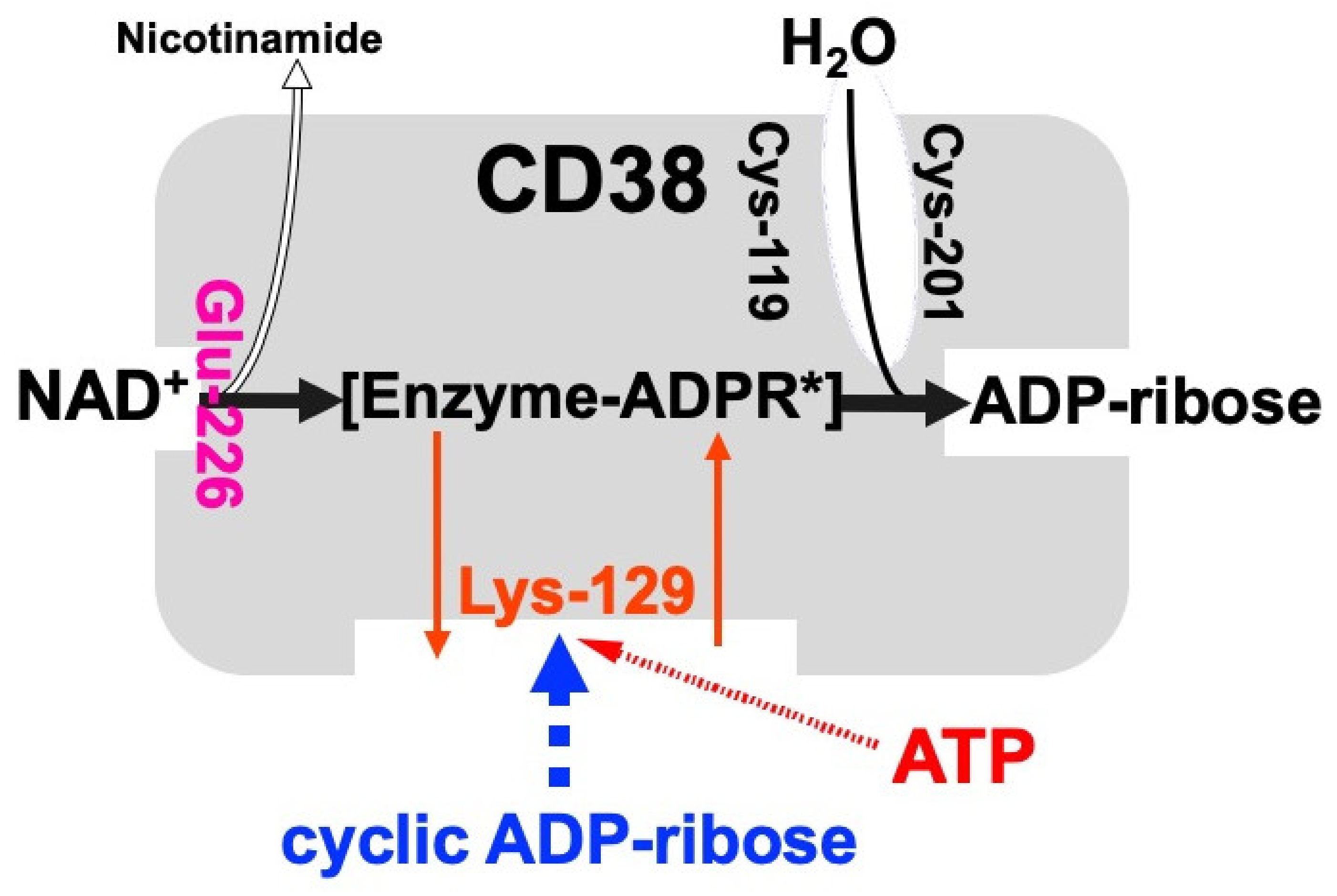
Figure 2. Schematic representation of human CD38 in enzyme activities for the synthesis and hydrolysis of cADPR (adapted from [26]). Glu-226 in human CD38 is essential for cADPR synthesis from NAD
Schematic representation of human CD38 in enzyme activities for the synthesis and hydrolysis of cADPR [26]. Glu-226 in human CD38 is essential for cADPR synthesis from NAD
+ (ADP-ribosyl cyclase activity) [46]. Cys-119 and Cys-201 are essential for cADPR hydrolysis to form ADP-ribose (cADPR hydrolase activity). Lys-129 is the cADPR binding site and is indispensable for cADPR hydrolysis (cADPR hydrolase) [21]. ATP, produced by glucose metabolism, competes with cADPR for the binding site (Lys-129), inhibiting the cADPR hydrolysis by cADPR hydrolase of CD38, which causes the accumulation of cADPR in the cell [12]. [Enzyme-cADPR*] is proposed as an enzyme-stabilized ADP-ribosyl oxocarbonium ion intermediate.
The human CD38 gene was assigned to band p15 of chromosome four as a single copy gene by in situ fluorescence hybridization [22]. The researchers then isolated the human CD38 gene and determined its primary structure [27]. The human CD38 gene as well as the Xenopus Cd38 gene extending ~70 kb [27,44] contained eight exons (
(ADP-ribosyl cyclase activity) [46]. Cys-119 and Cys-201 are essential for cADPR hydrolysis to form ADP-ribose (cADPR hydrolase activity). Lys-129 is the cADPR binding site and is indispensable for cADPR hydrolysis (cADPR hydrolase) [21]. ATP, produced by glucose metabolism, competes with cADPR for the binding site (Lys-129), inhibiting the cADPR hydrolysis by cADPR hydrolase of CD38, which causes the accumulation of cADPR in the cell [12]. [Enzyme-cADPR*] is proposed as an enzyme-stabilized ADP-ribosyl oxocarbonium ion intermediate.
The human CD38 gene was assigned to band p15 of chromosome four as a single copy gene by in situ fluorescence hybridization [22]. The researchers then isolated the human CD38 gene and determined its primary structure [27]. The human CD38 gene as well as the Xenopus Cd38 gene extending ~70 kb [27][44] contained eight exons (
Figure 3
). The human and
Xenopus Cd38
promoter regions have no TATA box but do have a CpG island, a methylation-controlled region more frequently associated with housekeeping than tissue-specific genes. Exon one encoded the 5′-untranslated region, translational start site, putative transmembrane domain and N-terminal region of Cd38. Exons two to eight encoded the remainder of Cd38. Cycsteines-119 and -201 of human CD38, which are indispensable for the hydrolysis of cADPR to form non-Ca
2+ mobilizing ADPR [21], were encoded in exons two and five, respectively. Lysine-129, which is involved in cADPR binding to CD38 [26], was encoded in exon three. Glutamate-226 and Trp-185, which seem to play important roles in catalytic activities [46,47], were encoded in exons six and four, respectively. The 10 cysteine residues conserved among human, rat, mouse, chicken, and frog Cd38s; human, rat, mouse, chicken, and frog Cd157s; Aplysia ADP-ribosyl cyclases, purple sea urchin ADP-ribosyl cyclase, and blood fluke ADP-ribosyl cyclase were encoded into six exons [27,32,44]. The translational termination codon and 3′-untranslated region were located in exon eight.
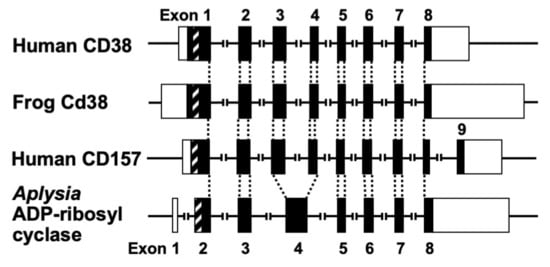
mobilizing ADPR [21], were encoded in exons two and five, respectively. Lysine-129, which is involved in cADPR binding to CD38 [26], was encoded in exon three. Glutamate-226 and Trp-185, which seem to play important roles in catalytic activities [46][47], were encoded in exons six and four, respectively. The 10 cysteine residues conserved among human, rat, mouse, chicken, and frog Cd38s; human, rat, mouse, chicken, and frog Cd157s; Aplysia ADP-ribosyl cyclases, purple sea urchin ADP-ribosyl cyclase, and blood fluke ADP-ribosyl cyclase were encoded into six exons [27][32][44]. The translational termination codon and 3′-untranslated region were located in exon eight.
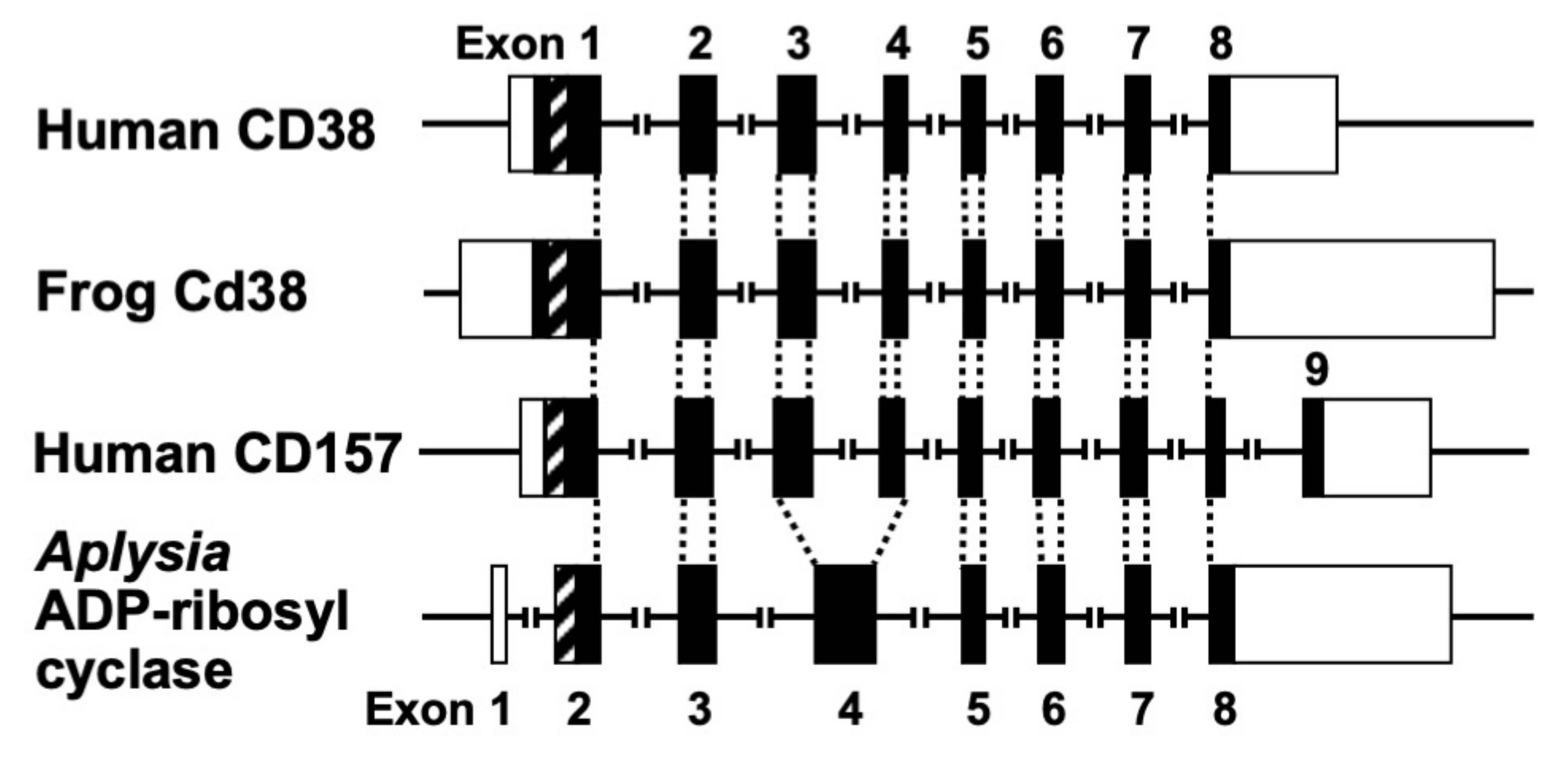
Figure 3. Structural organization of human CD38, frog Cd38, human CD157, and Aplysia kurodai ADP-ribosyl cyclase genes (adapted from [9,27,44]). The exons are depicted as boxes; filled and open boxes represent protein-coding regions and untranslated regions, respectively. Recently, primate-specific exon 1b, located between exon 1 and 2 of human CD157, that encodes 15 additional amino acids, was reported [48]. Hatched boxes represent transmembrane (CD38)- or signal peptide (CD157 and Aplysia ADP-ribosyl cyclase)-coding regions. The corresponding exon-intron junctions are indicated as broken lines. Exons are numbered.
Isolation and determination of the primary structure of the
Structural organization of human CD38, frog Cd38, human CD157, and Aplysia kurodai ADP-ribosyl cyclase genes [9][27][44]. The exons are depicted as boxes; filled and open boxes represent protein-coding regions and untranslated regions, respectively. Recently, primate-specific exon 1b, located between exon 1 and 2 of human CD157, that encodes 15 additional amino acids, was reported [48]. Hatched boxes represent transmembrane (CD38)- or signal peptide (CD157 and Aplysia ADP-ribosyl cyclase)-coding regions. The corresponding exon-intron junctions are indicated as broken lines. Exons are numbered.
Isolation and determination of the primary structure of the
Aplysia kurodai ADP-ribosyl cyclase gene [9] and the frog (Xenopus laevis) Cd38 gene [44] as well as the human CD38 gene [27] were performed. The exon–intron organization of the Aplysia ADP-ribosyl cyclase gene and the frog Cd38 gene is very similar to that of the human CD38 gene, suggesting that they evolved from a common ancestral gene [27,44] (
ADP-ribosyl cyclase gene [9] and the frog (Xenopus laevis) Cd38 gene [44] as well as the human CD38 gene [27] were performed. The exon–intron organization of the Aplysia ADP-ribosyl cyclase gene and the frog Cd38 gene is very similar to that of the human CD38 gene, suggesting that they evolved from a common ancestral gene [27][44] (
Figure 3
).
Kaisho et al. [49] found that the amino acid sequence of bone marrow stromal antigen 1 (BST-1/CD157) had significant sequence homology (~30% identity) with those of CD38 and Aplysia ADP-ribosyl cyclase. They determined the gene structure of human CD157 [50]. The gene extended for ~30 kb, very close to its paralogue CD38, and consisted of nine exons. Comparison of the gene organization reveals that the human CD157 gene has an exon–intron organization similar to that of human CD38 and Aplysia ADP-ribosyl cyclase genes (
Figure 3
). However, the human CD157 gene has an additional exon, exon nine, that encodes a peptide that is removed upon attachment of the glycosyl phosphatidylinositol anchor. In addition to the similar exon–intron structures of CD38, CD157 and Aplysia ADP-ribosyl cyclase genes and human CD38 and CD157 genes are located on the subregion of the human chromosome 4p15 as a next neighbor; the fact that mouse Cd157 (known as BP-3) is very close to the map site of the Cd38 gene on mouse chromosome 5 [51] strongly suggests that the three genes (CD38, CD157, and Aplysia ADP-ribosyl cyclase) have evolved from a common ancestor, and CD38 and CD157 genes were created by gene duplication before human and rodent divergence (
Figure 4).

).

Figure 4. Alignment of the amino acid sequences of the CD38/CD157/ADP-ribosyl cyclase family based on the primary structures of the encoded proteins (adapted from [44]). Dots indicate amino acids identical to human CD38. Dashes indicate gaps for maximal alignment. Ten cysteine and five amino acid residues conserved among all the proteins are highlighted in red and gray, respectively. The underlined amino acid residues are a putative transmembrane domain in CD38 and signal sequence in CD157 and ADP-ribosyl cyclase. The sequences used in alignment are #1: human CD38 (AAA68482 [55]), #2: rat CD38 (BAA06129 [20]), #3: mouse CD38 (AAA03163 [56]), #4: chicken CD38 (NM_001201388), #5: Xenopus laevis CD38 (AB194899 [44]), #6: human BST1 (CD157) (BAA04885 [47]), #7: rat BST1 (CD157) (Q63072 [51]), #8: mouse BST1 (CD157) (BAA06597 [57]), #9: chicken CD157 (NM_001200043), #10: Xenopus laevis CD157 (AB194901 [44]), #11: Aplysia californica ADP-ribosyl cyclase (AAA65698 [8]), #12: Aplysia kurodai ADP-ribosyl cyclase (BAA06284 [9]), #13: Strongylocentrotus purpuratus ADP-ribosyl cyclase (AM494973 [58]), and #14: Schistoma mansoni ADP-ribosyl cyclase (AAX35328 [59]). Although XP_005162637 (Danio rerio ADP-ribosyl cyclase 1-like [60]) showed significant homology with CD38/CD157/ADP-ribosyl cyclase family, the 8th and 10th cysteines in the conserved 10 cysteine residues (red), which are essential for the enzyme activities, are not conserved. It may be important to measure ADP-ribosyl cyclase activity in zebrafish ADP-ribosyl cyclase 1-like for understanding the involvement of the two cysteines in ADP-ribosyl cyclase activity.
The expression of Cd157 was reported in rat and mouse pancreatic β-cells [52,53]. However, no diabetic features were observed in Cd157 knockout mice [54]. Furthermore, CD157 exhibits neither cADPR-synthesizing (ADP-ribosyl cyclase) nor -hydrolyzing (cADPR hydrolase) activity in physiological conditions [14], suggesting that CD157 may mainly play a role as a surface antigen rather than a major enzyme for the synthesis of cADPR in response to glucose stimulation in pancreatic β-cells.
Alignment of the amino acid sequences of the CD38/CD157/ADP-ribosyl cyclase family based on the primary structures of the encoded proteins [44]. Dots indicate amino acids identical to human CD38. Dashes indicate gaps for maximal alignment. Ten cysteine and five amino acid residues conserved among all the proteins are highlighted in red and gray, respectively. The underlined amino acid residues are a putative transmembrane domain in CD38 and signal sequence in CD157 and ADP-ribosyl cyclase. The sequences used in alignment are #1: human CD38 (AAA68482 [52]), #2: rat CD38 (BAA06129 [20]), #3: mouse CD38 (AAA03163 [53]), #4: chicken CD38 (NM_001201388), #5: Xenopus laevis CD38 (AB194899 [44]), #6: human BST1 (CD157) (BAA04885 [47]), #7: rat BST1 (CD157) (Q63072 [51]), #8: mouse BST1 (CD157) (BAA06597 [54]), #9: chicken CD157 (NM_001200043), #10: Xenopus laevis CD157 (AB194901 [44]), #11: Aplysia californica ADP-ribosyl cyclase (AAA65698 [8]), #12: Aplysia kurodai ADP-ribosyl cyclase (BAA06284 [9]), #13: Strongylocentrotus purpuratus ADP-ribosyl cyclase (AM494973 [55]), and #14: Schistoma mansoni ADP-ribosyl cyclase (AAX35328 [56]). Although XP_005162637 (Danio rerio ADP-ribosyl cyclase 1-like [57]) showed significant homology with CD38/CD157/ADP-ribosyl cyclase family, the 8th and 10th cysteines in the conserved 10 cysteine residues (red), which are essential for the enzyme activities, are not conserved. It may be important to measure ADP-ribosyl cyclase activity in zebrafish ADP-ribosyl cyclase 1-like for understanding the involvement of the two cysteines in ADP-ribosyl cyclase activity.
The expression of Cd157 was reported in rat and mouse pancreatic β-cells [58][59]. However, no diabetic features were observed in Cd157 knockout mice [60]. Furthermore, CD157 exhibits neither cADPR-synthesizing (ADP-ribosyl cyclase) nor -hydrolyzing (cADPR hydrolase) activity in physiological conditions [14], suggesting that CD157 may mainly play a role as a surface antigen rather than a major enzyme for the synthesis of cADPR in response to glucose stimulation in pancreatic β-cells.