1. Introduction
The central dogma in genetics is that information in our cells flows only in one direction, from DNA to RNA, then to proteins. It was an absolute dogma that has now been essentially debunked due to the role of the environment in the modulation of gene expression
[1]. Environmental factors, including pollutants and lifestyle, constitute a significant role in severe, chronic pathologies with social and economic consequences
[2].
The measurement of all environmental exposures and assessing their correlation with effects on individual health is defined as the exposome
[3]. An individual’s exposome begins before birth and includes insults from environmental and occupational sources. In fact, the exposome interacts with our unique characteristics such as genetics, physiology, and epigenetics. The “environmental exposure” is a complex construct that encompasses exposures either physical, chemical, biological, or societal
[4]. Historically, Christopher Paul Wild defined the exposome in 2005 as the totality of an individual’s exposure experience from conception until death and its impact on chronic diseases
[5]. As a concept, it was put forward to stress the necessity of appropriate tools development for exposure assessment when applied to the study of human disease’s etiology
[6].
The adaptation of cells to environmental factors causing stress relies on a wide range of tightly controlled regulatory mechanisms. The epigenetic landscape represents the platform where multiple environmental factors interact with the complex genetic milieu, resulting in alterations in the expression gene that shape many aspects of health and disease
[7]. Changes in the organization and the structure of chromatin structure are associated with the transcriptional response to stress caused by the environment, and in some cases, can impart the memory of stress exposure to subsequent generations through mechanisms of epigenetic inheritance
[8].
Epigenetics investigates modifications in the expression of genes that do not depend on the underlying DNA sequence. Some studies have confirmed that environmental factors, such as toxicants, may promote a phenotype or a disease in an individual or even in the subsequent progeny through epigenetic alterations
[9,10][9][10]. Several epigenetic mechanisms, including modifications in DNA (e.g., methylation), histones, and non-protein coding RNAs (ncRNAs) can change genome expression under the exogenous influence
[11]. Notably, the role of long noncoding RNAs (lncRNAs) in epigenetic processes has recently been highlighted in more detail
[12]. The latent interest in epigenetics has resulted in breakthroughs in seminal concepts in diseases ranging from autoimmune conditions to cancer, congenital diseases, mental retardation, endocrine diseases, pediatric diseases, neuropsychiatric disorders, and many others
[1,13][1][13].
Discrepancies in the homeostatic functions of the epigenetic machinery cause a range of different disorders since these mechanisms (“epigenome”) are more sensitive to the environmental status (lack of nutrient consumption, physicochemical exposures, and psychological stress) than the genome, especially during early development due to the inherently dynamic nature of the epigenetic landscape
[14]. Besides the factors above, exposure to pollutants such as arsenic, nickel, cadmium, mercury, benzene, dioxin, bisphenol A, and diethylstilbestrol can alter the epigenetic regulation of the genome. These changes can cause abnormal gene expression, leading to various cancer types and other diseases
[15]. Here,
wthe
review current evidence indicating that epigenetic alterations mediate those detrimental effects caused by exposure to environmental toxicants, focusing mainly on a direct regulation by a diversity of ncRNAs subtypes (
Figure 1).
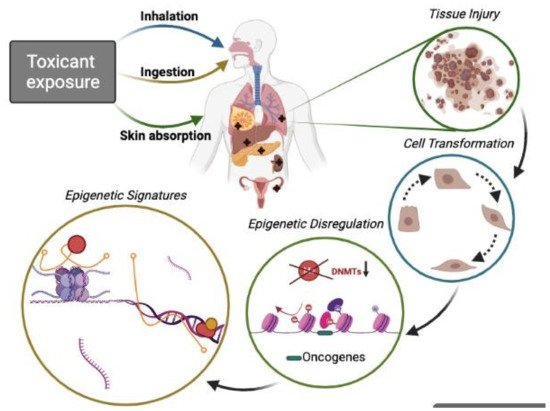
Figure 1. Epigenetic toxicants lead to the phenotypic transformation of normal cells. People exposed (by inhalation, food or water ingestion, and skin contact) to certain pollutants suffer potential tissue injury in the lung, mammary gland, liver, pancreas, skin, colon, ovary, and hematological tissue among other target organs. It is well known that acute or chronic exposures to toxicants are associated with malignant cell transformation and, thus, pollution-related diseases, including cancer. Aberrant genetic modifications (DNA methylation, histone modifications, and ncRNAs can transform cells and disturb the expression of genes involved in homeostasis maintenance. Recently much attention has been given to ncRNAs with their role in pathophysiological conditions and signaling pathways such as oncogenesis, cell survival, altered apoptosis, and cell adhesion. Thus, ncRNAs (miRNA, lncRNA) are vital mediator molecules. Moreover, other important regulators, such as ncRNA-associated proteins forming multi-component complexes on specific loci at specific time points that conform complex epigenetic signatures, are also crucial during the cell transformation process. Studying those epigenetic signatures may improve the understanding of the biology of different pollution-related cancer types.