Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Vicky Zhou and Version 3 by Vicky Zhou.
The structures composed of a pyridopyrimidine moiety which have shown a therapeutic interest or have already been approved for use as therapeutics, including pyrido[2,3-d]pyrimidines, pyrido[3,4-d]pyrimidines, pyrido[4,3-d]pyrimidines and pyrido[3,2-d]pyrimidines.
- pyridopyrimidines
- synthesis
- biological activity
- N-heterocycles
1. Introduction: Pyridopyrimidines and Their Scaffold
Depending on where the nitrogen atom is located in pyridine, it can be found four possible skeletons for the heterocyclic combination of pyrimidine and pyridine rings (Figure 1). Pyridopyrimidines and other N-heterocycles are of great interest due to their biological potential. The pyridopyrimidine moiety is present in relevant drugs and, in recent years, it has been studied in the development of new therapies, as evidenced by numerous publications, studies and clinical trials [1][2][3].
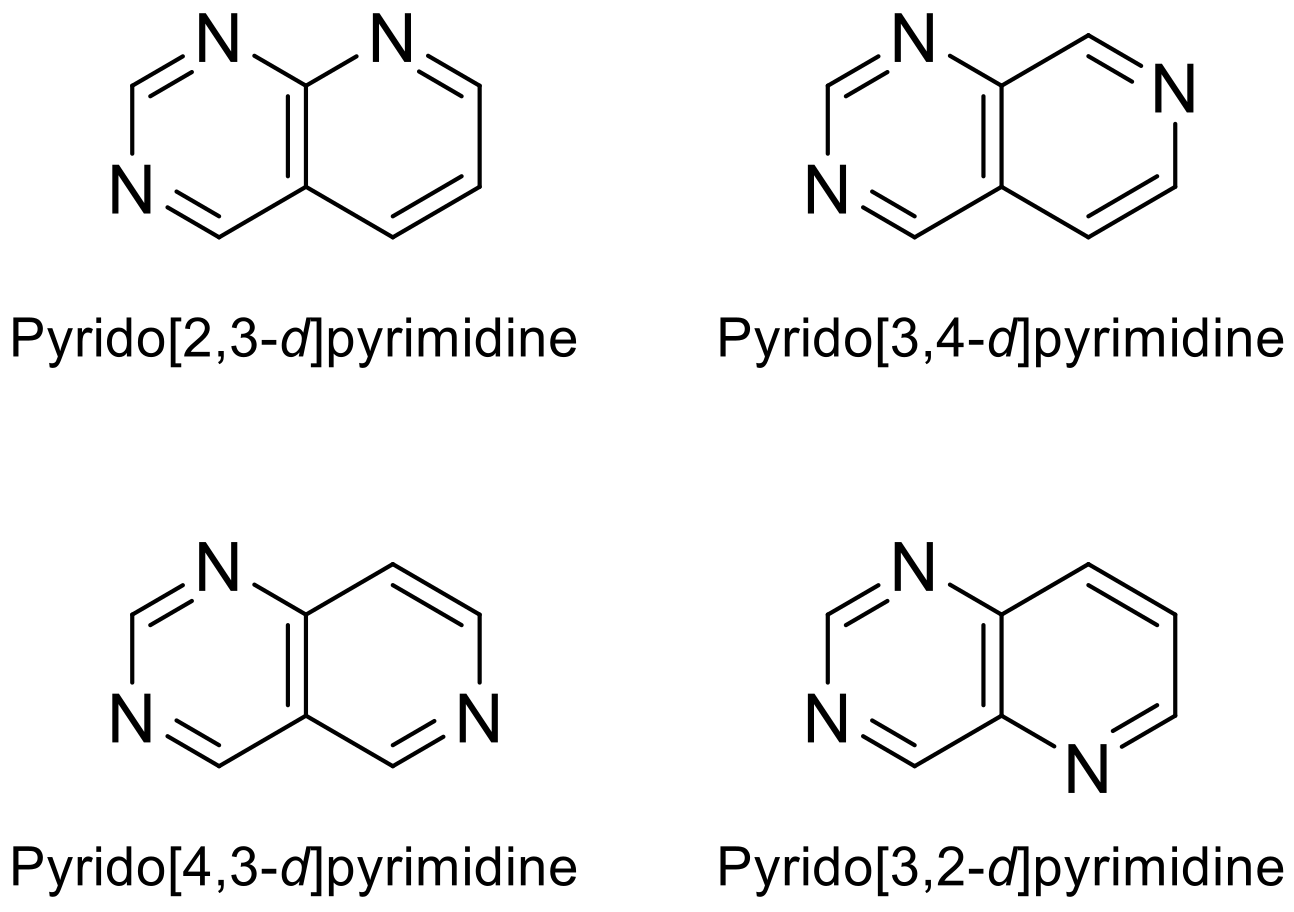
Figure 1. Various pyridopyrimidine structures types.
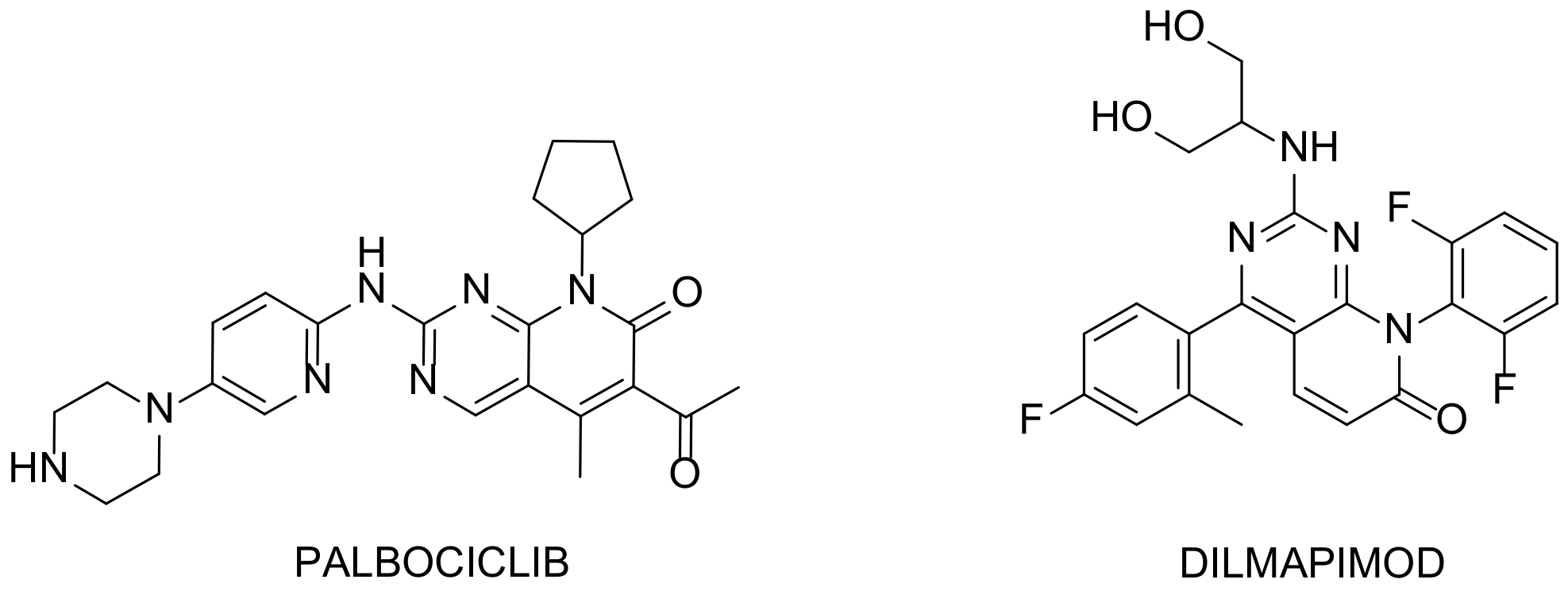
Figure 2. Examples of interesting molecules. Palbociclib: breast cancer drug developed by Pfizer and Dilmapimod: potential activity against rheumatoid arthritis.
2. Pyridopyrimidines: Therapeutic Potential and Synthesis
This section describes that, for each compound mentioned, the biological activity and the synthetic route reported. The 24 compounds described herein are presented according to the type of pyridopyrimidines (pyrido[2,3-d]pyrimidine, pyrido[3,4-d]pyrimidine, pyrido[4,3-d]pyrimidine and pyrido[3,2-d]pyrimidine). For each compound described, the target is indicated and some additional information has been added if different from that mentioned in the introduction.2.1. Pyrido[2,3-d]pyrimidine
Herein starts with some interesting pyrido[2,3-d]pyrimidines. The first one is 5-methyl-6-([methyl(3,4,5-trimethoxyphenyl)amino]methyl)pyrido[2,3-d]pyrimidine-2,4-diamine (Table 1, entry 1) which has been described to have DHFR dihydrofolate as the target [20].
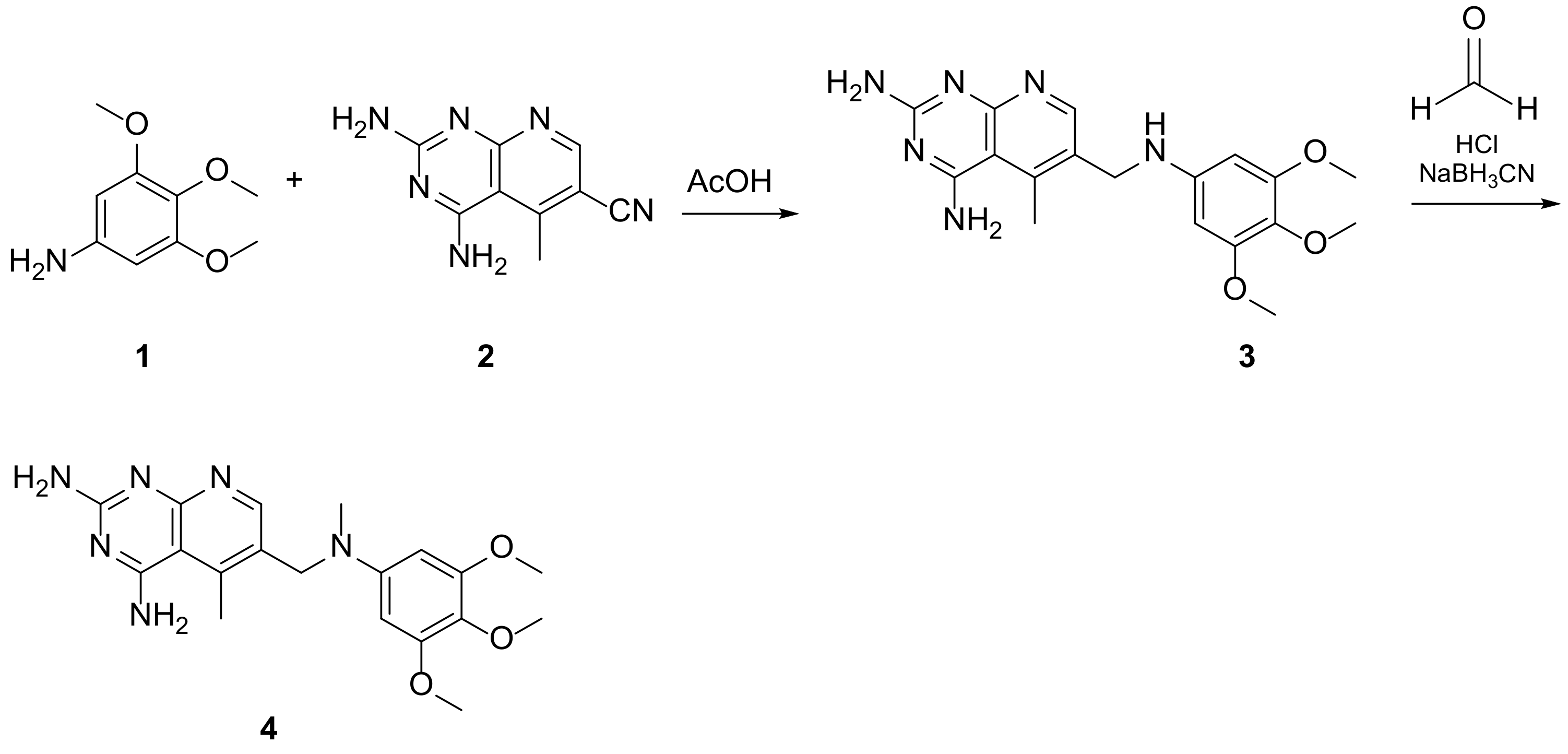
Kisliuk et al. described, in 1993, the synthesis of pyrido[2,3-d]pyrimidine-2,4-diamine (4). The reductive condensation of 6-cyano-5-methyl-pyrido[2,3-d]pyrimidine-2,4-diamine (2) with 3,4,5-trimethoxyaniline (1) in the presence of Raney Ni 70% in acetic acid gave the precursor 3 which underwent methylation at the N10 position by reductive alkylation with formaldehyde and sodium cyanoborohydride (Scheme 1) [20].
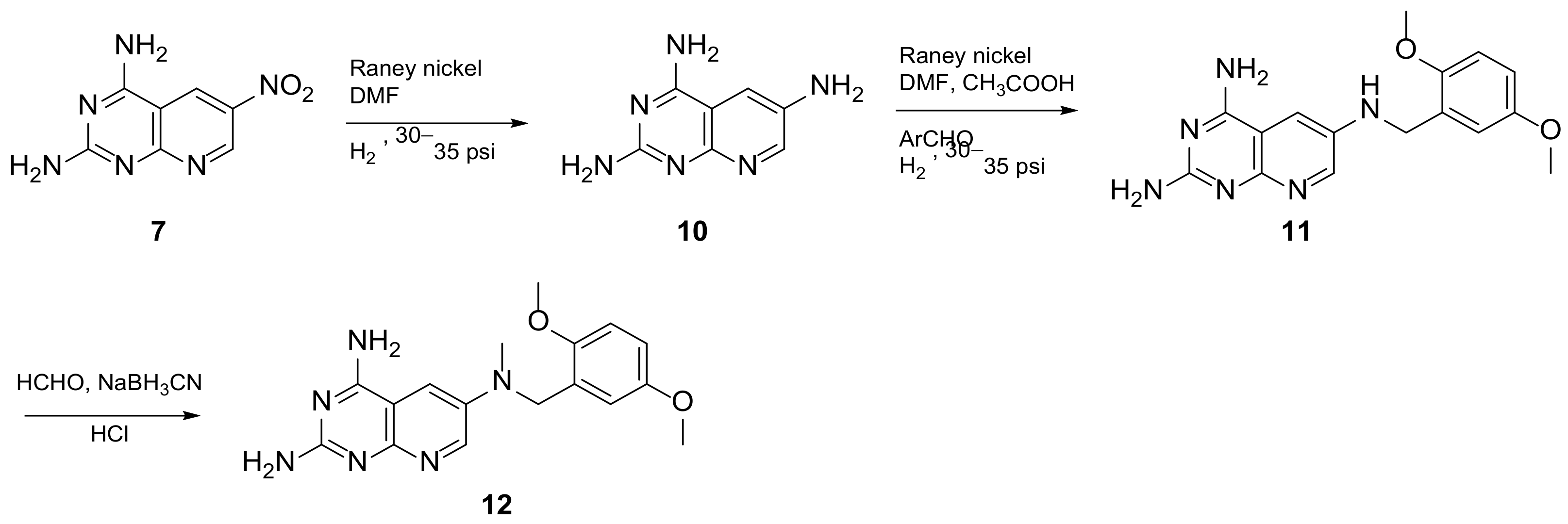
 In 2008, Queener et al. synthesized 12 starting from 2,4-diamino-6-nitroquinazoline 7 which underwent reduction with hydrogen and Raney nickel at 30-35 psi, providing the desired 2,4,6-triaminoquinazoline (10) (Scheme 3). Then, as described above, the 2,5-dimethoxybenzaldehyde ArCHO was added to generate the N9-H precursor 11. The following step was a reductive N9-alkylation using sodium cyanoborohydride which afforded the final compound [14]. Queener et al. conducted a biological evaluation of this compound 12 (Scheme 3, Table 1, entry 4) as a lipophilic inhibitor of dihydrofolate reductase.
In 2008, Queener et al. synthesized 12 starting from 2,4-diamino-6-nitroquinazoline 7 which underwent reduction with hydrogen and Raney nickel at 30-35 psi, providing the desired 2,4,6-triaminoquinazoline (10) (Scheme 3). Then, as described above, the 2,5-dimethoxybenzaldehyde ArCHO was added to generate the N9-H precursor 11. The following step was a reductive N9-alkylation using sodium cyanoborohydride which afforded the final compound [14]. Queener et al. conducted a biological evaluation of this compound 12 (Scheme 3, Table 1, entry 4) as a lipophilic inhibitor of dihydrofolate reductase.
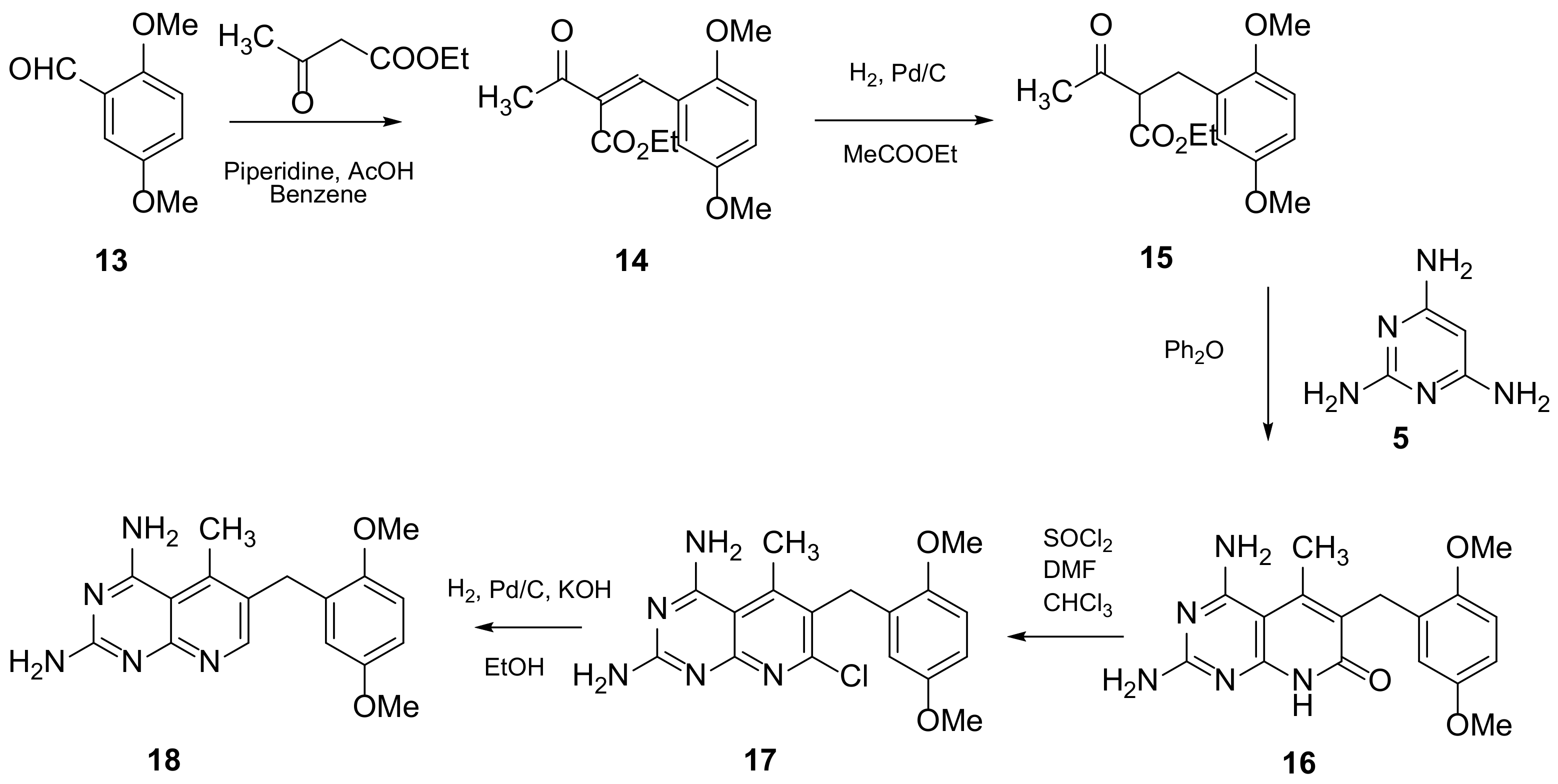
 Piritrexim (PTX) (Scheme 4 and Scheme 5, Table 1, entry 5) is a synthetic antifolate first synthesized by Grivsky, Sigel et al. [21] with anti-parasitic, anti-psoriatic and anti-tumor properties. Piritrexim inhibited dihydrofolate reductase (DHFR) and also showed good antitumor effects on the carcinosarcoma in rats. An advantage of this compound compared to some analogues is that it does not have effects as an inhibitor of histamine metabolism, reducing the potential risk of side reactions on metabolism. Its degree of lipophilicity, i.e., the affinity of this drug for a lipid environment, allows it to diffuse easily into the cells. The various therapeutical activities listed for piritrexim are on melanoma and urothelial cancer, and promising results in head and neck cancer were already obtained in combination with other molecules [16].
Piritrexim (PTX) (Scheme 4 and Scheme 5, Table 1, entry 5) is a synthetic antifolate first synthesized by Grivsky, Sigel et al. [21] with anti-parasitic, anti-psoriatic and anti-tumor properties. Piritrexim inhibited dihydrofolate reductase (DHFR) and also showed good antitumor effects on the carcinosarcoma in rats. An advantage of this compound compared to some analogues is that it does not have effects as an inhibitor of histamine metabolism, reducing the potential risk of side reactions on metabolism. Its degree of lipophilicity, i.e., the affinity of this drug for a lipid environment, allows it to diffuse easily into the cells. The various therapeutical activities listed for piritrexim are on melanoma and urothelial cancer, and promising results in head and neck cancer were already obtained in combination with other molecules [16].
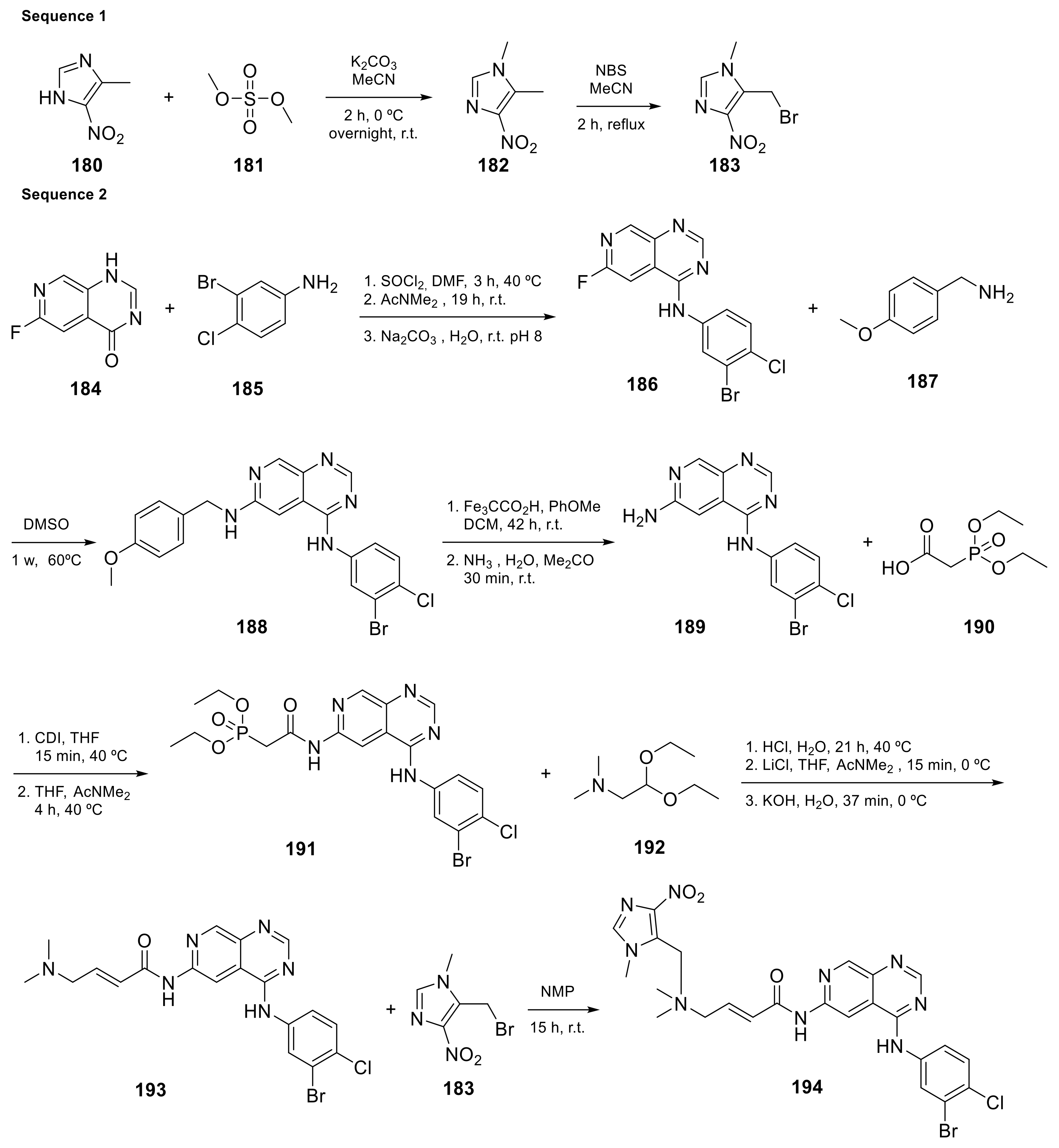 Carlin et al. [23] patented in 2015 the preparation of 4-anilinopyrido[3,4-d]pyrimidine prodrugs (Scheme 6, Table 1, entry 19) as kinase inhibitors useful for cancer treatment. The procedure is described in Scheme 6 with classical synthetic methodologies affording the expected compound 194 in twelve steps.
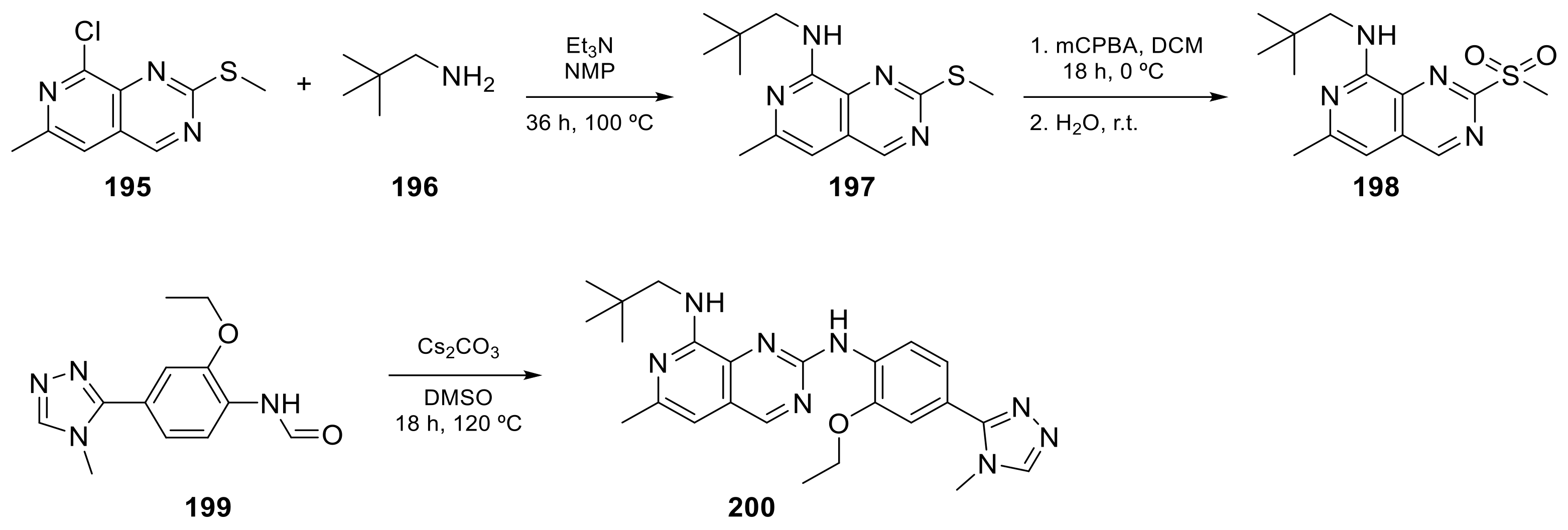
The second example is the BOS172722 derivative (200 in Scheme 7, Table 1, entry 20). This compound, in combination with paclitaxel, was tested in vivo for the treatment of triple hormone receptor-negative breast cancer demonstrating a promising synergy. This selective monopolar spindle 1 (Mps1) kinase inhibitor has been identified as a potential anti-cancer agent because it is involved in the division of cancer cells. This is, therefore, an attractive target for cancer therapy [24][25]. It has the dual specificity protein kinase TTK as the target.
Carlin et al. [23] patented in 2015 the preparation of 4-anilinopyrido[3,4-d]pyrimidine prodrugs (Scheme 6, Table 1, entry 19) as kinase inhibitors useful for cancer treatment. The procedure is described in Scheme 6 with classical synthetic methodologies affording the expected compound 194 in twelve steps.
The second example is the BOS172722 derivative (200 in Scheme 7, Table 1, entry 20). This compound, in combination with paclitaxel, was tested in vivo for the treatment of triple hormone receptor-negative breast cancer demonstrating a promising synergy. This selective monopolar spindle 1 (Mps1) kinase inhibitor has been identified as a potential anti-cancer agent because it is involved in the division of cancer cells. This is, therefore, an attractive target for cancer therapy [24][25]. It has the dual specificity protein kinase TTK as the target.
Scheme 1. Synthesis of pyrido[2,3-d]pyrimidine-2,4-diamine (4) by Kisliuk et al. [20].
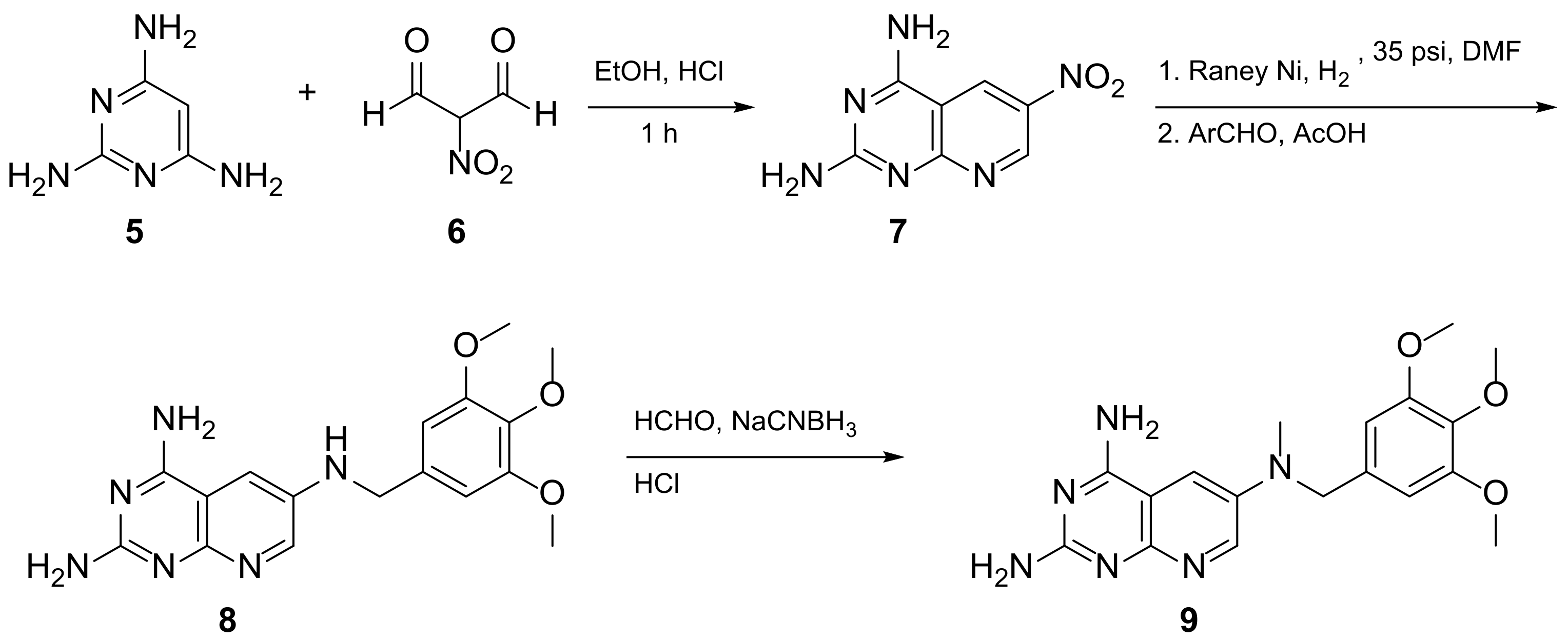
Kisliuk et al. also developed another strategy to synthesize pyrido[2,3-d] pyrimidine-2,4-diamines as compound 9 (Scheme 2, Table 1, entry 2). Starting from 2,4,6-triaminopyrimidine (5) with the sodium salt of nitromalonaldehyde, they obtained in a single step the 2,4-diamino-6-nitropyrido [2,3-d]pyrimidine (7) which was then reduced to its corresponding 6-amino analogue using Raney Ni in DMF. The reductive amination with various aldehydes (ArCHO, in this case 3,4,5-trimethoxybenzaldehyde) provided the desired product 8. In the last step, 8 was N-methylated by treatment with formaldehyde in the presence of sodium cyanoborohydride [15] (Scheme 2). An analog compound (Table 1, entry 3) was obtained following the same synthetic pathway (Scheme 2) using 3,5-dimethoxybenzaldehyde.
Scheme 2. Synthesis of pyrido[2,3-d]pyrimidine-2,4-diamine (9) by Kisliuk et al. [15].
Scheme 3. Synthesis of N6-[(2,5-dimethoxyphenyl)methyl]-N6-methylpyrido[2,3-d]pyrimidine-2,4,6-triamine (12) by Queener et al. [14].
Scheme 4. Synthesis of 6-[(2,5-dimethoxyphenyl)methyl]-5-methylpyrido[2,3-d]pyrimidine-2,4-diamine (18) by Grivsky, Sigel et al. [21].
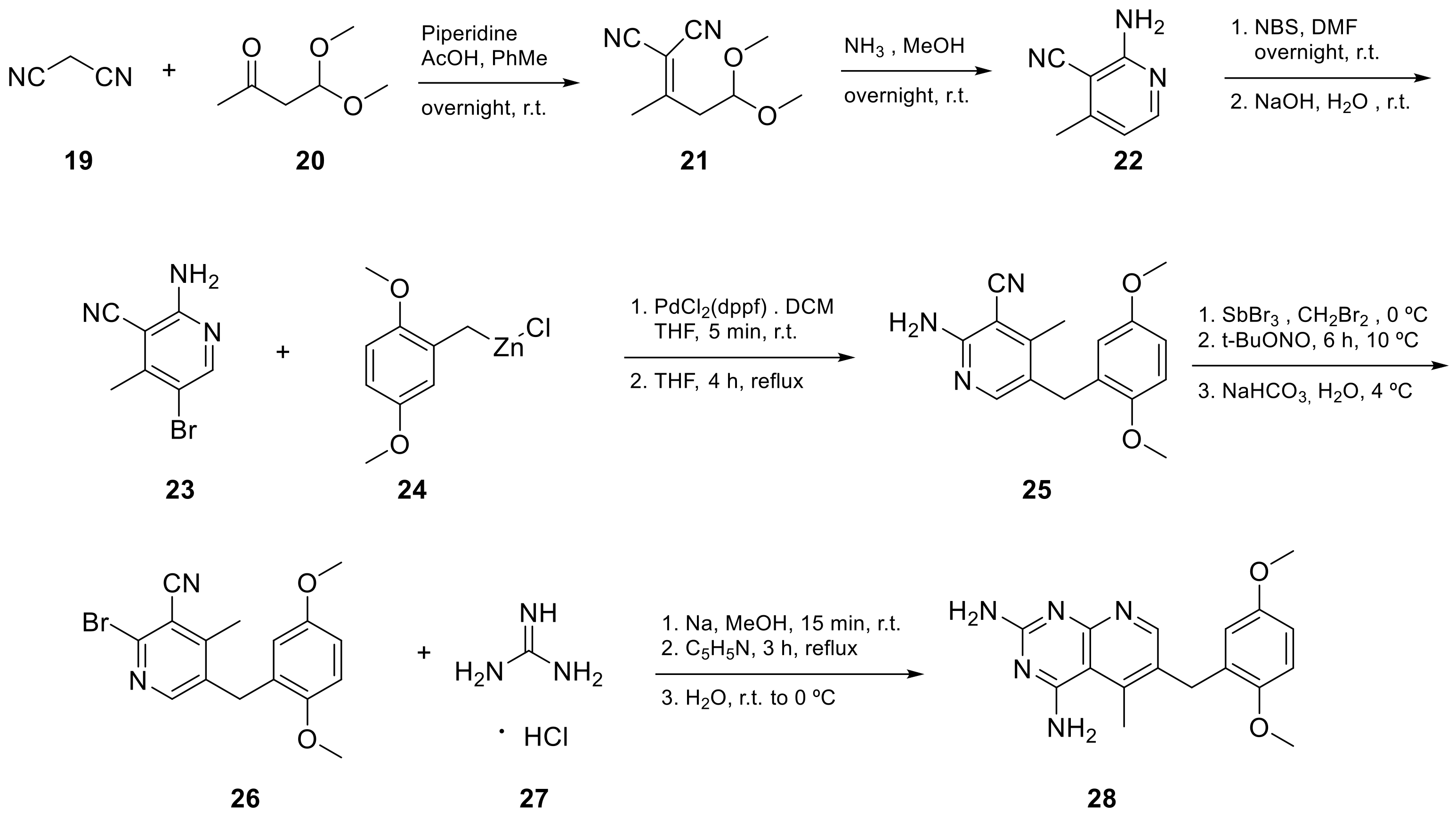
Scheme 5. Synthesis of 6-[(2,5-dimethoxyphenyl)methyl]-5-methylpyrido[2,3-d]pyrimidine-2-amine (28) by Chan and Rosowsky [17].
2.2. Pyrido[3,4-d]pyrimidine
This class of pyridopyrimidine is mainly referenced with kinase activity. The first example mentioned herein is Tarloxotinib (194 in Scheme 6). It is being studied in the clinical trial NCT03743350 (NSCLC exon 20 or HER2 activating mutation) [22]. This molecule is a kinase inhibitor targeting all members of the HER family, with a novel mechanism of action. It is a hypoxia-activated prodrug that releases an active metabolite irreversibly targeting the kinase. The goal is to inhibit only HER kinases in tumor cells. Tarloxotinib is a Pan-HER kinase inhibitor.
Scheme 6. [(2E)-3-({4-[(3-bromo-4-chlorophenyl)amino]pyrido[3,4-d]pyrimidin-6-yl}carbamoyl)prop-2-en-1-yl]dimethyl[(1-methyl-4-nitro-1H-imidazol-5-yl)methyl]azanium (194) [23].
Scheme 7.
N
8-(2,2-dimethylpropyl)-
N
2-[2-ethoxy-4-(4-methyl-4
H
-1,2,4-triazol-3-yl)phenyl]-6-methylpyrido[3,4-
d
]pyrimidine-2,8-diamine (
200
), BOS172722
[24]
.
2.3. Pyrido[4,3-d]pyrimidine
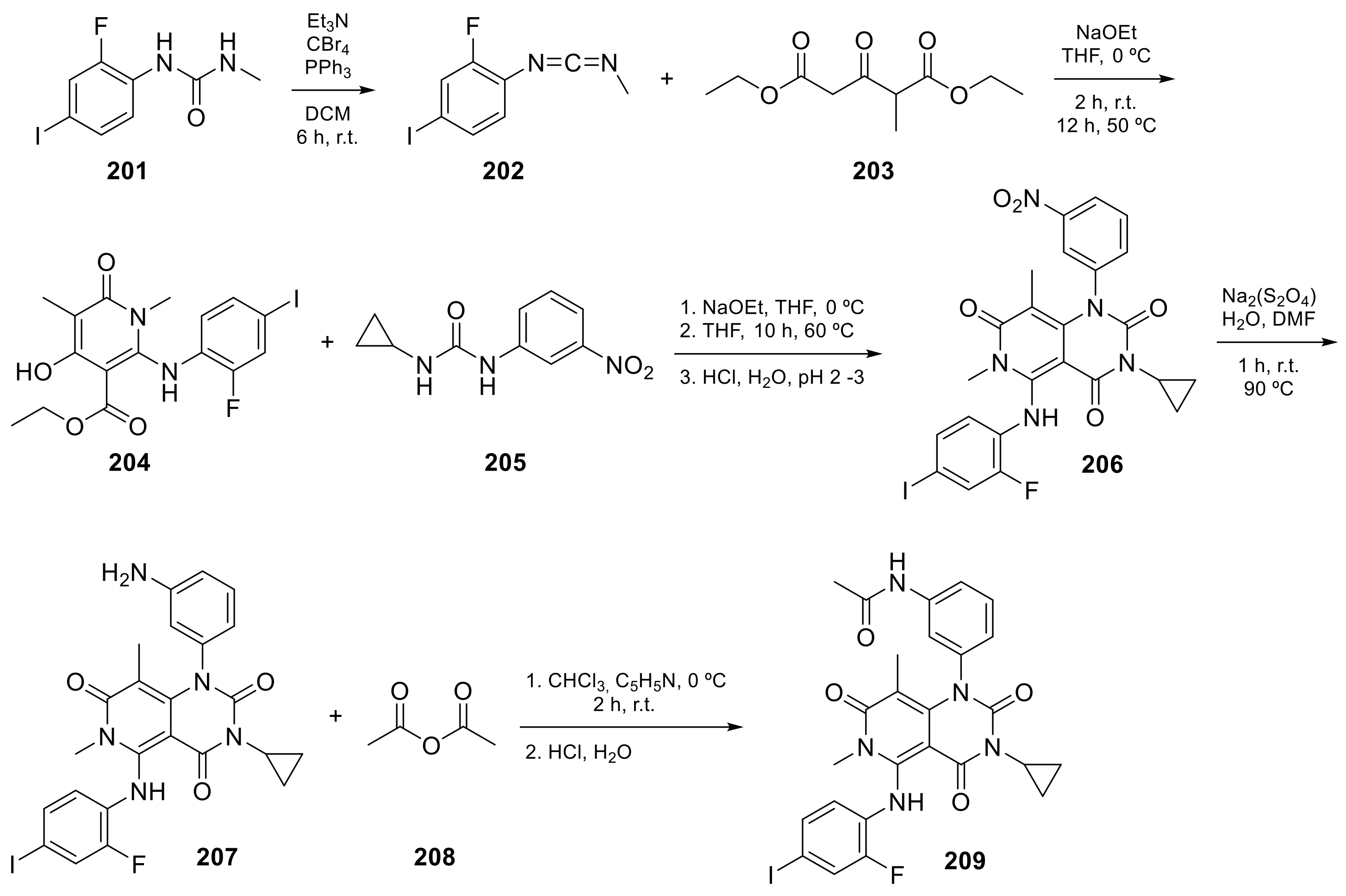
Trametinib (209 in Scheme 8, Table 1, entry 21) is a kinase inhibitor used for specific types of melanoma. This compound, associated with other molecules such as Dabrafenib (Tafilnar) and/or Mekinist (trametinib), has been approved by the FDA in particular for the treatment of degenerative thyroid cancer (ATC) [26][27].
Scheme 8. Synthesis of N-(3-(3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7-trioxo-1H,2H,3H,4H,6H,7H-pyrido[4,3-d]pyrimidin-1-yl)phenyl)acetamid, Trametinib (209) [28].
2.4. Pyrido[3,2-d]pyrimidine
Seletalisib (229 in Scheme 9, Table 1, entry 22) is a novel small-molecule inhibitor of PI3Kδ that was evaluated in clinical assays to study the treatment and basic science of Primary Sjogren’s Syndrome [29]. This molecule is an ATP-competitive and highly selective PI3Kδ inhibitor. Phosphoinositide 3-kinases (PI3K) are enzymes regulating cellular survival, development, and function. They play a key role in immune cell development and function.
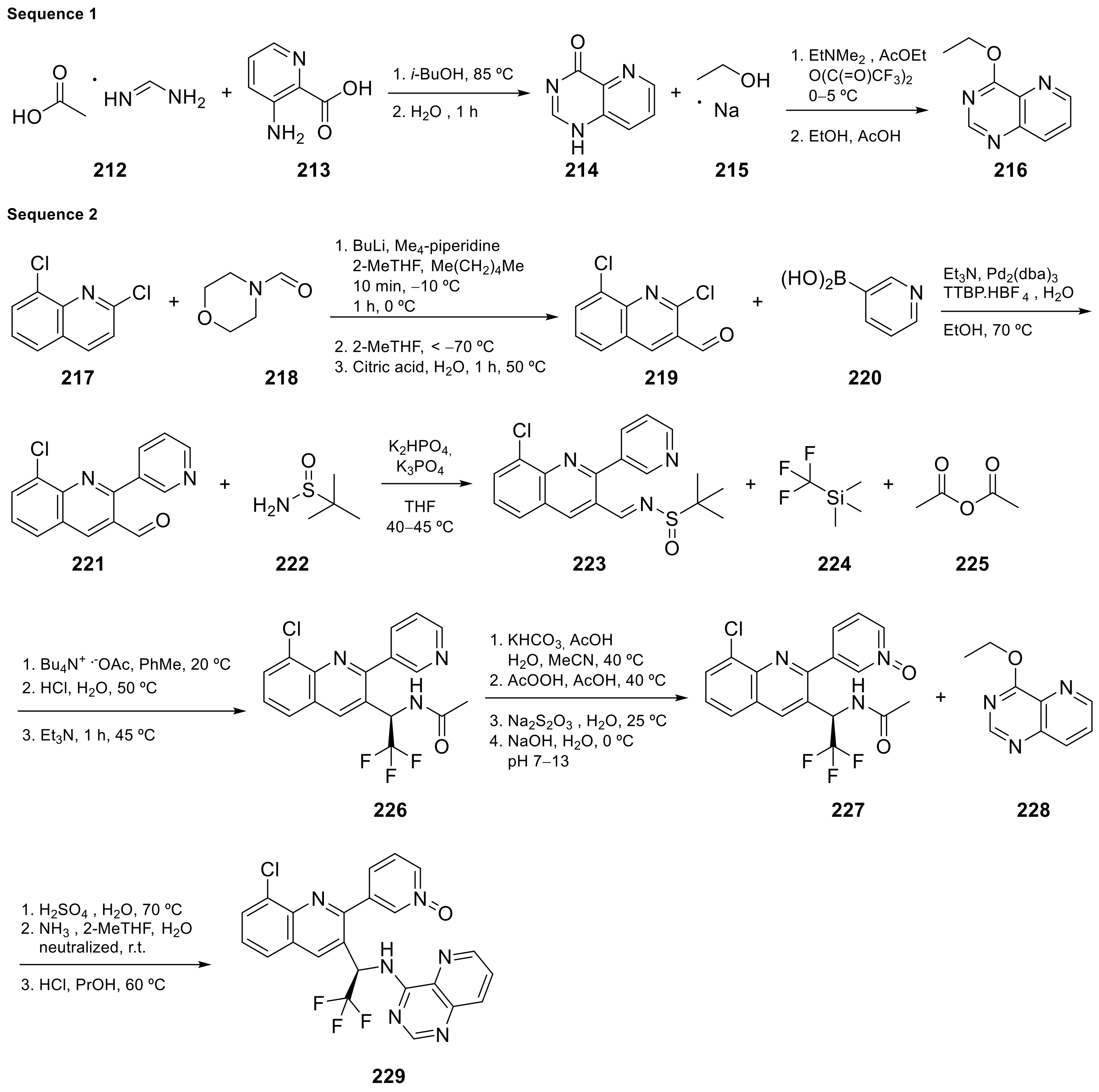
Scheme 9.
Synthesis of 3-[8-chloro-3-[(1
R
)-2,2,2-trifluoro-1-([pyrido[3,2-
d
]pyrimidin-4-yl]amino)ethyl]quinolin-2-yl]pyridin-1-ium-1-olate (
229
)
[30]
.
Le Meur et al. [30] patented the synthesis of Seletalisib 229 following the procedure summarized in Scheme 9, and described crystalline forms for the treatment of various pathologies.
All compounds described herein with their target were listed below (Table 1).
Table 1. The 24 pyridopyrimidines described here.
| Entry | Structure | Name | Target | Ref. |
|---|---|---|---|---|
| Pyrido[2,3-d]pyrimidine | ||||
| 1 | 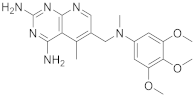 |
5-methyl-6-([methyl(3,4,5-trimethoxyphenyl)amino]methyl)pyrido[2,3-d]pyrimidine-2,4-diamine | DHFR Dihydrofolate reductase |
[31][32][20] |
| 2 | 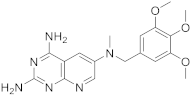 |
N6-methyl-N6-[(3,4,5-trimethoxyphenyl)methyl]pyrido[2,3-d]pyrimidine-2,4,6-triamine | DHFR Dihydrofolate reductase |
[15] |
| 3 | 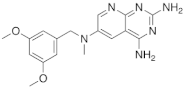 |
N6-[(3,5-dimethoxyphenyl)methyl]-N6-methylpyrido[2,3-d]pyrimidine-2,4,6-triamine, | DHFR Dihydrofolate reductase |
[15] |
| 4 | 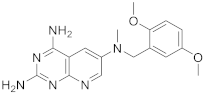 |
N6-[(2,5-dimethoxyphenyl)methyl]-N6-methylpyrido[2,3-d]pyrimidine-2,4,6-triamine | DHFR Dihydrofolate reductase |
[14] |
| 5 | 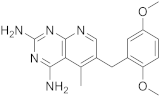 PIRITREXIM |
6-[(2,5-dimethoxyphenyl)methyl]-5-methylpyrido[2,3-d]pyrimidine-2,4-diamine | DHFR Dihydrofolate reductase |
[16][17][21] |
| 6 |  |
6-(2,6-dichlorophenyl)-2-([3-(hydroxymethyl)phenyl]amino)-8-ethyl-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Tyrosine kinase activity | [33][34] |
| 7 | 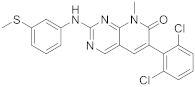 PD-173955 |
6-(2,6-dichlorophenyl)-8-methyl-2-([3-(methylsulfanyl)phenyl]amino)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: Tyrosine-protein kinase transforming protein Abl |
[13] |
| 8 | 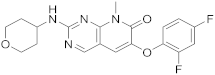 |
6-(2,4-difluorophenoxy)-8-methyl-2-[(oxan-4-yl)amino]-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: Mitogen-activated protein kinase 14 |
[35][36][37] |
| 9 | 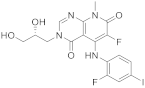 TAK-733 |
3-[(2R)-2,3-dihydroxypropyl]-6-fluoro-5-[(2-fluoro-4-iodophenyl)amino]-8-methyl-3H,4H,7H,8H-pyrido[2,3-d]pyrimidine-4,7-dione | Kinase activity: Against MEK and ERK |
[38][39][40][41] |
| 10 | 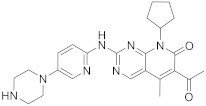 Palbociclib |
6-acetyl-8-cyclopentyl-5-methyl-2-([5-(piperazin-1-yl)pyridin-2-yl]amino)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: Cyclin-dependent kinase 4/Cyclin-dependent kinase 6 Breast cancer drug |
[42][43][44][45][46] |
| 11 | 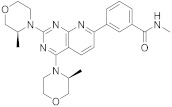 Vistusertib |
3-(2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl)-N-methylbenzamide | Kinase activity: Vistusertib (AZD2014) is a novel mTOR inhibitor |
[47][48] |
| 12 | 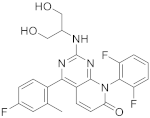 Dilmapimod (SB-681323) |
8-(2,6-difluorophenyl)-2-[(1,3-dihydroxypropan-2-yl)amino]-4-(4-fluoro-2-methylphenyl)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: P38 MAPK inhibitor, Tumor necrosis factor/Interleukin-1 beta/Interleukin-6. Potential activity against rheumatoid arthritis | [49][50][51][52][53][54][55] |
| 13 | 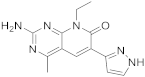 Voxtalisib |
2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: PI3K/mTOR Inhibitor |
[56][57] |
| 14 | 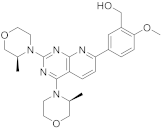 AZD8055 |
(5-(2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl)-2-methoxyphenyl)methanol | Kinase activity: Selective ATP-competitive mTOR kinase inhibitor. Induction of MEK/ERK |
[58][59] |
| 15 | 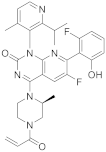 AMG-510 |
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]-1H,2H-pyrido[2,3-d]pyrimidin-2-one | Kinase Activity: KRAS inhibitor implicated in the RAS/MAPK pathway GTPase KRas |
[60][61][62][63][64] |
| 16 | 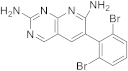 |
6-(2,6-dibromophenyl)pyrido[2,3-d]pyrimidine-2,7-diamine | Biotin carboxylase | [65] |
| 17 | 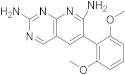 |
6-(2,6-dimethoxyphenyl)pyrido[2,3-d]pyrimidine-2,7-diamine | Biotin carboxylase | [33][66] |
| 18 | 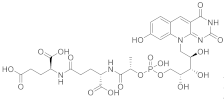 |
(2S)-2-[(4S)-4-carboxy-4-[(2S)-2-([hydroxy(([(2R,3S,4S)-2,3,4-trihydroxy-5-(8-hydroxy-2,4-dioxo-2H,3H,4H,10H-pyrimido[4,5-b]quinolin-10-yl)pentyl]oxy))phosphoryl]oxy)propanamido]butanamido]pentanedioic acid | Methanobacterium redox coenzyme Factor 420 (F420) | [67] |
| Pyrido[3,4-d]pyrimidine | ||||
| 19 | 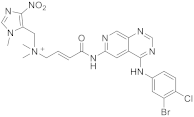 Tarloxotinib |
[(2E)-3-((4-[(3-bromo-4-chlorophenyl)amino]pyrido[3,4-d]pyrimidin-6-yl)carbamoyl)prop-2-en-1-yl]dimethyl[(1-methyl-4-nitro-1H-imidazol-5-yl)methyl]azanium | Kinase Activity: Pan-HER kinase inibitor |
[23] |
| 20 | 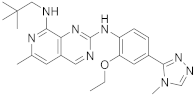 BOS172722 |
N8-(2,2-dimethylpropyl)-N2-[2-ethoxy-4-(4-methyl-4H-1,2,4-triazol-3-yl)phenyl]-6-methylpyrido[3,4-d]pyrimidine-2,8-diamine | Kinase Activity: Dual specificity protein kinase TTK |
[24][25][68][69] |
| Pyrido[4,3-d]pyrimidine | ||||
| 21 | 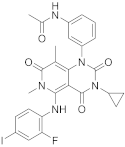 Trametinib |
N-(3-(3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7-trioxo-1H,2H,3H,4H,6H,7H-pyrido[4,3-d]pyrimidin-1-yl)phenyl)acetamide | Dual specificity mitogen-activated protein kinase kinase 1/Dual specificity mitogen-activated protein kinase kinase 2 | [26][27][70][71] |
| Pyrido[3,2-d]pyrimidine | ||||
| 22 | 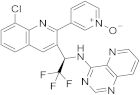 Seletalisib |
3-(8-chloro-3-[(1R)-2,2,2-trifluoro-1-((pyrido[3,2-d]pyrimidin-4-yl)amino)ethyl]quinolin-2-yl)pyridin-1-ium-1-olate | selective PI3Kδ inhibitor | [72][30] |
| 23 | 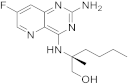 |
(2S)-2-((2-amino-7-fluoropyrido[3,2-d]pyrimidin-4-yl)amino)-2-methylhexan-1-ol | Chronic hepatitis B TLR8 receptor |
[73][74] |
| 24 | 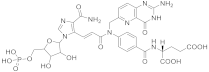 β-DADF |
(2S)-2-((4-[(2E)-N-((2-amino-4-oxo-1H,4H-pyrido[3,2-d]pyrimidin-6-yl)methyl)-3-(4-carbamoyl-1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-[(phosphonooxy)methyl]oxolan-2-yl]-1H-imidazol-5-yl)prop-2-enamido]phenyl)formamido)pentanedioic acid | Bifunctional purine biosynthesis protein PURH | [75][76] |
References
- Yadav, P.; Shah, K. An overview on synthetic and pharmaceutical prospective of pyridopyrimidines scaffold. Chem. Biol. Drug Des. 2021, 3, 633–648.
- Yousif, M.N.M.; El-Gazzar, A.-R.B.A.; El-Enany, M.M. Synthesis and Biological Evaluation of Pyridopyrimidines. Mini Rev. Org. Chem. 2021, 1, 43–54.
- Buron, F.; Mérour, J.Y.; Akssira, M.; Guillaumet, G.; Routier, S. Recent advances in the chemistry and biology of pyridopyrimidines. Eur. J. Med. Chem. 2015, 95, 76–95.
- Cao, X.; Tanis, K.Q.; Koleske, A.J.; Colicelli, J. Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J. Biol. Chem. 2008, 283, 31401–31407.
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264.
- Segalés, J.; Perdiguero, E.; Muñoz-Cánoves, P. Regulation of muscle stem cell functions: A focus on the p38 MAPK Signaling Pathway. Front. Cell Dev. Biol. 2016, 4, 91.
- Orton, R.J.; Sturm, O.E.; Vyshemirsky, V.; Calder, M.; Gilbert, D.R.; Kolch, W. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochem. J. 2005, 392, 249–261.
- Wakil, S.J.; Stoops, J.K.; Joshi, V.C. Fatty acid synthesis and its regulation. Ann. Rev. Biochem. 1983, 52, 537–579.
- Smejkalova, H.; Erb, T.J.; Fuchs, G. Methanol assimilation in methylobacterium extorquens AM1: Demonstration of all enzymes and their regulation. PLoS ONE 2010, 5, e13001.
- Erb, T.J.; Berg, I.A.; Brecht, V.; Muller, M.; Fuchs, G.; Alber, B.E. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 10631–10636.
- Khomyakova, M.; Bukmez, O.; Thomas, L.K.; Erb, T.J.; Berg, I.A. A methylaspartate cycle in haloarchaea. Science 2011, 331, 334–337.
- Alber, B.E. Biotechnological potential of the ethylmalonyl-CoA pathway. Appl. Microbiol. Biotechnol. 2011, 89, 17–25.
- Heinzlmeir, S.; Kudlinzki, D.; Sreeramulu, S.; Klaeger, S.; Gande, S.L.; Linhard, V.; Wilhelm, M.; Qiao, H.; Helm, D.; Ruprecht, B.; et al. Chemical proteomics and structural biology define EPHA2 inhibition by clinical kinase drugs. ACS Chem. Biol. 2016, 11, 3400–3411.
- Gangjee, A.; Adair, O.O.; Pagley, M.; Queener, S.F. N9-substituted 2,4-diaminoquinazolines: Synthesis and biological evaluation of lipophilic inhibitors of pneumocystis carinii and toxoplasma gondii dihydrofolate reductase. J. Med. Chem. 2008, 51, 6195–6200.
- Gangjee, A.; Vasudevan, A.; Queener, S.F.; Kisliuk, R.L. 2,4-Diamino-5-deaza-6-substituted pyridopyrimidine antifolates as potent and selective nonclassical inhibitors of dihydrofolate reductases. J. Med. Chem. 1996, 39, 1438–1446.
- Piper, J.R.; Johnson, C.A.; Krauth, C.A.; Carter, R.L.; Hosmer, C.A.; Queener, S.F.; Borotz, S.E.; Pfefferkorn, E.R. Lipophilic antifolates as agents against opportunistic infections. 1. Agents superior to trimetrexate and piritrexim against toxoplasma gondii and pneumocystis carinii in vitro evaluations. J. Med. Chem. 1996, 39, 1271–1280.
- Chan, D.C.M.; Rosowsky, A. Synthesis of the lipophilic antifolate piritrexim via a palladium(0)-catalyzed cross-coupling reaction. J. Org. Chem. 2005, 70, 1364–1368.
- Tong, L. Structure and function of biotin-dependent carboxylases. Cell. Mol. Life Sci. 2013, 5, 863–891.
- Shivaiah, K.-K.; Upton, B.; Nikolau, B.J. Kinetic, structural, and mutational analysis of Acyl-CoA carboxylase from thermobifida fusca YX. Front. Mol. Biosci. 2021, 7, 615614.
- Gangjee, A.; Shi, J.; Queener, S.F.; Barrows, L.R.; Kisliuk, R.L. Synthesis of 5-methyl-5-deaza nonclassical antifolates as inhibitors of dihydrofolate reductases and as potential antipneumocystis, antitoxoplasma, and antitumor agents. J. Med. Chem. 1993, 36, 3437–3443.
- Grivsky, E.M.; Lee, S.; Sigel, C.W.; Duch, D.S.; Nichol, C.A. Synthesis and antitumor activity of 2,4-diamino-6-(2,5-dimethoxybenzyl)-5-methylpyridopyrimidine. J. Med. Chem. 1980, 23, 327–329.
- Estrada-Bernal, A.; Le, A.T.; Doak, A.E.; Tirunagaru, V.G.; Silva, S.; Bull, M.R.; Smaill, J.B.; Patterson, A.V.; Kim, C.; Liu, S.V.; et al. Tarloxotinib Is a Hypoxia-Activated Pan-HER kinase inhibitor active against a broad range of HER-family oncogenes. Clin. Cancer Res. 2021, 5, 1463–1475.
- Smaill, J.B.; Patterson, A.V.; Lu, G.-L.; Lee, H.H.; Ashoorzadeh, A.; Anderson, R.F.; Wilson, W.R.; Denny, W.A.; Hsu, H.-L.A.; Maroz, A.; et al. Preparation of 4-Anilinopyridopyridine Prodrugs as Kinase Inhibitors Useful for Treatment of Cancer. U.S Patent No. 9101632 B2, 11 August 2015.
- Woodward, H.L.; Innocenti, P.; Cheung, K.-M.J.; Hayes, A.; Roberts, J.; Henley, A.T.; Faisal, A.; Mak, G.W.-Y.; Box, G.; Westwood, I.M.; et al. Introduction of a methyl group curbs metabolism of pyridopyrimidine monopolar spindle 1 (MPS1) inhibitors and enables the discovery of the phase 1 clinical candidate N2-(2-ethoxy-4-(4-methyl-4 H-1,2,4-triazol-3-yl)phenyl)-6-methyl-N8-neopentylpyridopyrimidine-2,8-diamine (BOS172722). J. Med. Chem. 2018, 18, 8226–8240.
- Anderhub, S.J.; Mak, G.W.-Y.; Gurden, M.D.; Faisal, A.; Drosopoulos, K.; Walsh, K.; Woodward, H.L.; Innocenti, P.; Westwood, I.M.; Naud, S.; et al. High proliferation rate and a compromised spindle assembly checkpoint confers sensitivity to the MPS1 inhibitor BOS172722 in triple-negative breast cancers. Mol. Cancer Ther. 2019, 10, 1696–1707.
- Zeiser, R.; Andrlová, H.; Meiss, F. Trametinib (GSK1120212) recent results. Cancer Res. 2018, 211, 91–100.
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 19, 1813–1823.
- Wu, X.; Hai, W.; Shi, Z. Method for Synthesizing Trametinib for Treating Melanoma with Low Cost. CN Patent No. 109320513 A, 12 February 2019.
- Juarez, M.; Diaz, N.; Johnston, G.I.; Nayar, S.; Payne, A.; Helmer, E.; Cain, D.; Williams, P.; Devauchelle-Pensec, V.; Fisher, B.A.; et al. A phase 2 randomized, double-blind, placebo-controlled, proof-of-concept study of oral seletalisib in primary Sjögren’s syndrome. Rheumatology 2021, 3, 1364–1375.
- Aerts, L.L.J.J.; Assaf, G.; Carly, N.E.; Cool, V.A.C.; Delatinne, J.-P.; Delhaye, L.J.W.; Kestemont, J.P.; Le Meur, S. Crystalline Forms of Seletalisib for Treatment of Inflammatory, Autoimmune, Cardiovascular, Neurodegenerative, Metabolic, Oncolologic, Nociceptive or Ophthalmic Conditions. WO Patent No. 2018219772 A1, 6 December 2018.
- Schweitzer, B.I.; Dicker, A.P.; Bertino, J.R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 8, 2441–2452.
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 6, 1140.
- Blankley, C.J.; Boschelli, D.H.; Doherty, A.M.; Hamby, J.M.; Klutchko, S.; Panek, R.L. Preparation of Pyridopyrimidines for Inhibiting Protein Tyrosine Kinase Mediated Cellular Proliferation. U.S. Patent No. 5733914 A, 31 March 1998.
- Klutchko, S.R.; Hamby, J.M.; Boschelli, D.H.; Wu, Z.; Kraker, A.J.; Amar, M.; Hartl, B.G.; Shen, C.; Klohs, W.D.; Steinkampf, R.W.; et al. 2-Substituted aminopyrido-pyrimidin-7(8H)-ones. Structure-Activity Relationships against selected tyrosine kinases and in vitro and in vivo anticancer activity. J. Med. Chem. 1998, 41, 3276–3292.
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849.
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 2, 153–183.
- Keri, G.; Oerfi, L.; Greff, Z.; Banhegyi, P.; Szantai-Kis, C.; Eros, D.; Zsakai, L.; Boros, S.; Breza, N. Novel Compounds as Kinase Inhibitors and Their Use for the Regulation of Fibrotic Cell Proliferation. HU Patent Application No. 2015000620 A2, 28 July 2017.
- Adjei, A.A.; LoRusso, P.; Ribas, A.; Sosman, J.A.; Pavlick, A.; Dy, G.K.; Zhou, X.; Gangolli, E.; Kneissl, M.; Faucette, S.; et al. A phase I dose-escalation study of TAK-733, an investigational oral MEK inhibitor, in patients with advanced solid tumors. Investig. New Drugs. 2017, 1, 47–58.
- Dong, Q.; Dougan, D.R.; Gong, X.; Halkowycz, P.; Jin, B.; Kanouni, T.; O’Connell, S.M.; Scorah, N.; Shi, L.; Wallace, M.B.; et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett. 2011, 21, 1315–1319.
- Micel, L.N.; Tentler, J.J.; Tan, A.C.; Selby, H.M.; Brunkow, K.L.; Robertson, K.M.; Lindsey Davis, S.; Klauck, P.J.; Pitts, T.M.; Gangolli, E.; et al. Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol. Cancer Ther. 2015, 14, 317–325.
- Zhao, Y.; Zhu, L.; Provencal, D.P.; Miller, T.A.; O’Bryan, C.; Langston, M.; Shen, M.; Bailey, D.; Sha, D.; Palmer, T.; et al. Process research and kilogram synthesis of an investigational, potent MEK inhibitor. Org. Process Res. Dev. 2012, 16, 1652–1659.
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016, 20, 1925–1936.
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Bartlett, C.H.; Zhang, K.; et al. PALOMA3 study group palbociclib in hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2015, 3, 209–219.
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2019, 2, 118–136.
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 15, 3079–3093.
- Chu, D.; Zhang, Y.; Xie, X.; Fang, L. Preparation of Palbociclib Intermediate and Method for Synthesizing Palbociclib. CN Patent No. 111675660 A, 2020.
- Lee, J.; Kim, S.T.; Kim, K.; Lee, H.; Kozarewa, I.; Mortimer, P.G.S.; Odegaard, J.I.; Harrington, E.A.; Lee, J.; Lee, T.; et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial. Cancer Discov. 2019, 10, 1388–1405.
- Shen, G.; Liu, M.; Lu, J.; Meng, T. Practical synthesis of vistusertib (AZD2014), an ATP competitive mTOR inhibitor. Tetrahedron Lett. 2019, 60, 151333.
- Anand, P.; Shenoy, R.; Palmer, J.E.; Baines, A.J.; Lai, R.Y.K.; Robertson, J.; Bird, N.; Ostenfeld, T.; Chizh, B.A. Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur. J. Pain. 2011, 10, 1040–1048.
- Christie, J.D.; Vaslef, S.; Chang, P.K.; May, A.K.; Gunn, S.R.; Yang, S.; Hardes, K.; Kahl, L.; Powley, W.M.; Lipson, D.A.; et al. A randomized dose-escalation study of the safety and anti-inflammatory activity of the p38 mitogen-activated protein kinase inhibitor dilmapimod in severe trauma subjects at risk for acute respiratory distress syndrome. Crit. Care Med. 2015, 9, 1859–1869.
- Ma, K.; Zhang, H.; Baloch, Z. Pathogenetic and therapeutic applications of tumor necrosis factor-α (TNF-α) in major depressive disorder: A systematic review. Int. J. Mol. Sci. 2016, 5, 733.
- Vanamee, E.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 511, eaao4910.
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-1 cytokine family--Balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 1, 25–37.
- Rose-John, S. Interleukin-6 family cytokines. Cold Spring Harb. Perspect. Biol. 2018, 2, a028415.
- Adams, J.L.; Boehm, J.C.; Hall, R.; Jin, Q.; Kasparec, J.; Silva, D.J.; Taggart, J.J. Preparation of 2,4,8-Trisubstituted-8H-pyridopyrimidin-7-ones as CSBP/RK/p38 Kinase Inhibitors. WO Patent No. 2002059083 A2, 1 August 2002.
- Mehnert, J.M.; Edelman, G.; Stein, M.; Camisa, H.; Lager, J.; Dedieu, J.-F.; Ghuysen, A.-F.; Sharma, J.; Liu, L.; LoRusso, P.M. A phase I dose-escalation study of the safety and pharmacokinetics of a tablet formulation of voxtalisib, a phosphoinositide 3-kinase inhibitor, in patients with solid tumors. Investig. New Drugs 2018, 1, 36–44.
- Cai, T.; Zaks, T.; Romanelli, A. Compositions and Methods for Treating Cancer Using PI3K Inhibitor and Anti-CD19 Maytansinoid Immunoconjugate. WO Patent No. 2014058947 A1, 17 April 2014.
- Xu, D.-Q.; Toyoda, H.; Qi, L.; Morimoto, M.; Hanaki, R.; Iwamoto, S.; Komada, Y.; Hirayama, M. Induction of MEK/ERK activity by AZD8055 confers acquired resistance in neuroblastoma. Biochem. Biophys. Res. Commun. 2018, 3, 425–432.
- Pike, K.G.; Malagu, K.; Hummersone, M.G.; Menear, K.A.; Duggan, H.M.E.; Gomez, S.; Martin, N.M.B.; Ruston, L.; Pass, S.L.; Pass, M. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: The discovery of AZD8055 and AZD2014. Bioorg. Med. Chem. Lett. 2013, 23, 1212–1216.
- Kettle, J.G.; Cassar, D.J. Covalent inhibitors of the GTPase KRAS G12C: A review of the patent literature. Expert. Opin. Ther Pat. 2020, 2, 103–120.
- Yang, H.; Xiang, S.; Kazi, A.; Sebti, S.M. The GTPase KRAS suppresses the p53 tumor suppressor by activating the NRF2-regulated antioxidant defense system in cancer cells. J. Biol. Chem. 2020, 10, 3055–3063.
- Wu, H.-Z.; Xiao, J.-Q.; Xiao, S.-S.; Cheng, Y. KRAS: A promising therapeutic target for cancer treatment. Curr. Top. Med. Chem. 2019, 23, 2081–2097.
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable cancer target. Cancer Treat. Rev. 2020, 89, 102070.
- Parsons, A.T.; Cochran, B.M.; Powazinik, W., IV; Caporini, M.A. Improved Synthesis of Key Intermediates of KRAS G12C Inhibitor. WO Patent No. 2020102730 A1, 22 May 2020.
- Skedelj, V.; Arsovska, E.; Tomasic, T.; Kroflic, A.; Hodnik, V.; Hrast, M.; Bester-Rogac, M.; Anderluh, G.; Gobec, S.; Bostock, J.; et al. 6-Arylpyrido pyrimidines as novel ATP-competitive inhibitors of bacterial D-alanine: D-alanine ligase. PLoS ONE 2012, 7, e39922.
- Blankley, C.J.; Doherty, A.M.; Hamby, J.M.; Panek, R.L.; Schroeder, M.C.; Showalter, H.D.H.; Connolly, C. Preparation of 6-Arylpyridopyrimidines and Naphthyridines for Inhibiting Protein Tyrosine Kinase Mediated Cellular Proliferation. U.S. Patent No. 5733913 A, 31 March 1998.
- Kimachi, T.; Kawase, M.; Matsuki, S.; Tanaka, K.; Yoneda, F. First total synthesis of coenzyme factor 420. J. Chem. Soc. Perkin Trans. 1 1990, 2, 253–256.
- Wang, S.; Zhang, M.; Liang, D.; Sun, W.; Zhang, C.; Jiang, M.; Liu, J.; Li, J.; Li, C.; Yang, X.; et al. Molecular design and anticancer activities of small-molecule monopolar spindle 1 inhibitors: A medicinal chemistry perspective. Eur. J. Med. Chem. 2019, 175, 247–268.
- Kessler, A.F.; Feldheim, J.; Schmitt, D.; Feldheim, J.J.; Monoranu, C.M.; Ernestus, R.-I.; Löhr, M.; Hagemann, C. Monopolar spindle 1 kinase (MPS1/TTK) mRNA expression is associated with earlier development of clinical symptoms, tumor aggressiveness and survival of glioma patients. Biomedicines 2020, 7, 192.
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 8, 1263–1284.
- Wu, X.; Hai, W.; Shi, Z. Method for Synthesizing Trametinib for Treating Melanoma with Low Cost. CN Patent No. 109320513 A, 12 February 2019.
- Juarez, M.; Diaz, N.; Johnston, G.I.; Nayar, S.; Payne, A.; Helmer, E.; Cain, D.; Williams, P.; Devauchelle-Pensec, V.; Fisher, B.A.; et al. A phase 2 randomized, double-blind, placebo-controlled, proof-of-concept study of oral seletalisib in primary Sjögren’s syndrome. Rheumatology 2021, 3, 1364–1375.
- Mackman, R.L.; Mish, M.; Chin, G.; Perry, J.K.; Appleby, T.; Aktoudianakis, V.; Metobo, S.; Pyun, P.; Niu, C.; Daffis, S.; et al. Discovery of GS-9688 (Selgantolimod) as a potent and selective oral toll-like receptor 8 agonist for the treatment of chronic hepatitis B. J. Med. Chem. 2020, 63, 10188–10203.
- Asselin, S.M.; Badalov, P.R.; Morrison, H.G.; Regens, C.S.; Vieira, T. Preparation of Solid Forms of (R)-2-pyrimidin-4-yl)amino]-2-methylhexan-1-ol as Toll-Like Receptor Modulators. WO Patent No. 2020214663 A1, 22 October 2020.
- Wall, M.; Shim, J.H.; Benkovic, S.J. A multisubstrate adduct inhibitor of AICAR transformylase. J. Med. Chem. 1999, 42, 3421–3424.
- Warren, M.S.; Mattia, K.M.; Marolewski, A.E.; Benkovic, S.J. The transformylase enzymes of de novo purine biosynthesis. Pure Appl. Chem. 1996, 68, 2029–2036.
More