Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 1 by Rita Moretti.
Small vessel disease (SVD) is one of the most frequent pathological conditions which lead to dementia. Biochemical and neuroimaging might help correctly identify the clinical diagnosis of this relevant brain disease. The microvascular alterations which underlie SVD have common origins, similar cognitive outcomes, and common vascular risk factors.
- small vessel disease
- vascular damage
- reactive oxygen species
- metalloproteinases
1. Possible and Proved New Markers of Blood–Brain Barrier Leakage, Perivascular Enlargements, and Mitochondrial Alterations
Small vessel disease (SVD) has the small vessels (pial and the small penetrating) and white matter as a significant definite target. Nevertheless, growing attention has been dedicated to disrupting perivascular spaces, astrocytic end-feet, capillaries, and veins. As a final point, the blood–brain barrier (BBB) has been addressed as another potential target of the intrigued mechanisms that underlie the small vessel brain pathology complex. BBB is not only a solid defensive barrier but acts as an active and specific player of active selection crossover, possessing cell-cell signaling with the end-feet of astrocytes and disclosure a potential role of maintaining efflux pumps [51,52,53,54,55][1][2][3][4][5]. Thus, the disruption of the BBB is proportionately increased by normal aging but progresses as a hallmark in different pathologies, i.e., multiple sclerosis or in primary inflammatory disease. Nevertheless, it is an expression of white matter inflammation, even due to chronic hypoperfusion, such as the one which occurs in small vessel disease [SVD], accomplishing the progression and the extension of the white matter sufferance, named as white matter hyperintensities (WMH) [56[6][7][8][9][10][11][12],57,58,59,60,61,62], the confluency of which is synonymous with SVD progression, leading to subcortical vascular dementia (sVAD) [12,62][13][12]. In AD-prone patients, BBB disruption has been signaled even in hippocampal degeneration, which occurs after a major stroke [61,62][11][12].
A dynamic contrast-enhanced MRI (DCE-MRI) [63][14] has been employed for in-vivo quantification of the pathological passage of plasma through BBB [64,65][15][16]. Moreover, apart from the BBB leakage, the possibility of estimating the vascular permeability-surface area product (PS) and the plasma volume fraction (VP) in a given region of interest has also been described [66,67][17][18]. The model suggested that PS increased with WMH severity, aging, and other vascular risk factors, and at the same time, a lower blood vP [65][16]. The most promising in-vivo demonstration is that BBB integrity is compromised in more severe WMH, even beyond visible lesions [63][14] (Insert Figure 1).
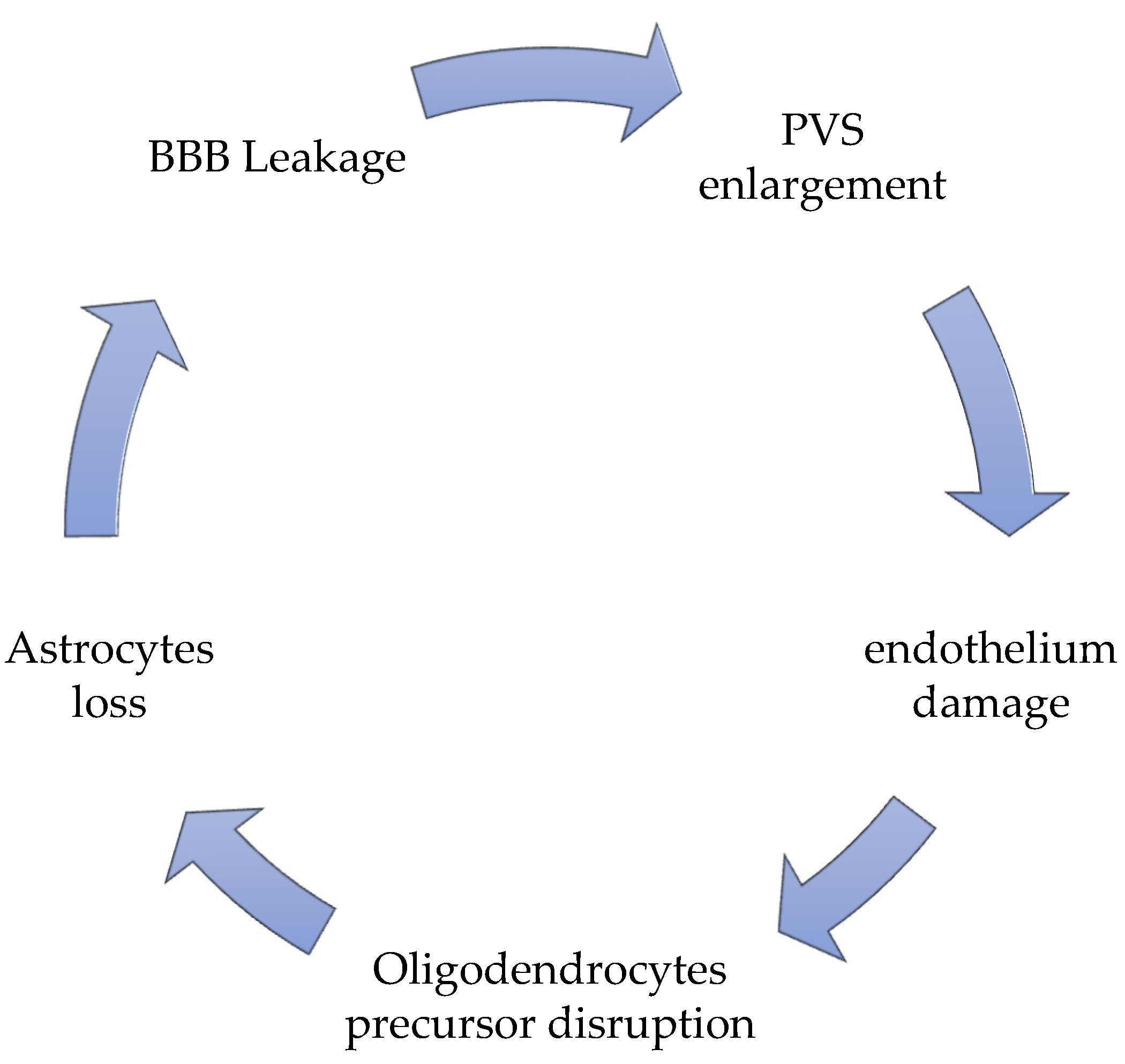
Figure 1.
The vicious circle of SVD pathology.
Even if it we knowas known that BBB is disrupted in SVD, we do not know thit is still unknown that the reasons for BBB leakage in this condition. The most disputed involvement is one of the pericytes. Pericytes are capillary mural cells that stabilize newly formed vessels and induce repair. When a pericyte-deficient adult mouse model has been employed [68][19], different transcriptional changes in brain endothelial cells have been mapped due to a defective pericyte contact at a single-cell level. In that conformation, endothelial cells, deprived of pericyte contacts, seem to exhibit a “venous-shifted molecular pattern,” and therefore lack any capillary specialization, and upregulate proteins which are typically expressed during developmental stages, such as the Fibroblast Growth Factor Binding Protein (Fgfbp1), or those expressed during pathological angiogenesis, such as Angiopoietin 2 (Angpt2). These aspects permit a possible cell proliferation, with a very flawed arteriolar BBB regulation system, and reduction of the angiogenesis process [68][19]. Fgfbp1 and Angpt2 levels could probably be crucial markers of BBB leakage during SVD. More studies will be necessary to prove that.
Perivascular spaces (PVS) have gained an essential role in SVD pathogenesis; they are no longer considered as virtual empty spaces, but as the most efficacious catabolites clearance system [12][13]; they are resident sites of perivascular macrophages, pial cells, mast cells, nerve fibers, and collagen fibers [69][20]. PVS are virtual spaces intimately connected to deep arterioles [70][21]. Even in these conditions, they act as a lymphatic net, defined as a glymphatic-perivascular territory [71][22].
Their malfunction, the hallmarks of which are the combined enlargement and widening, is the principal responsibility for perivascular accumulation of catabolites and toxic substances, which is determinant for enhancing ongoing neural damage until starvation [72,73][23][24]. The perivascular debris accumulation, together with the BBB leakage, potentiates and accelerates the perivascular inflammation, strongly favored by the stagnation-induced process and by medical conditions which influence it, such as hypertension and diabetes [74,75,76,77,78][25][26][27][28][29]. PVS enlargement is responsible for an altered cerebrovascular reactivity (CVR) [12][13], due to the extension of the constant inflammatory response [41][30] present as a constant marker in SVD, due to the chronic hypoperfusion state. The PVS is never an isolated situation, but it is accompanied by an altered BBB disruption and a significant perivascular inflammation [75,79,80,81,82][26][31][32][33][34]. More recently, new actors contribute with BBB leakage and PVS enlargement to help the progress of SVD [83[35][36][37][38],84,85,86], such as the oligodendrocyte precursor cells (OPCs), which generally help BBB stabilization [86,87][38][39] and the astrocytes, which exert their fundamental role as regulating the signal of neuro-vascular coupling [12][13]. Oligodendrocytes are the first victims of chronic models of chronic cerebral hypoperfusion (CCH), together with the precocious sufferance of the perineural space [88,89,90][40][41][42], and with a hyper-activation of microglia, firstly in the hippocampus [91[43][44],92], then in the thalamus, up to in the cortical neuronal population [93][45]. Secondary to oligodendrocytes, astrocyte death occurs in proportion to the chronic ischemia condition’s length and severity [94[46][47],95], due to the ongoing modifications of general and neuronal metabolic requests. Their death is a consequence of chronic hypoxia, but it worsens neuronal death due to a lack of functions, regulating the neurovascular coupling signal [96][48]. The process by which this occurs is that during the entire process of chronic ischemia, microglia retract its branches, with a consequent reduction of the length and strength of the microglial ramification, with a concomitant degeneration of the soma [97][49]. The frontal activation of microglia occurs in a two-step pattern: at the beginning, M1 activation upregulates TNF alpha, Il-23, IL-1beta, and Il12 production, which attack neurons, and directly contribute to their injury; only after M2 activation occurs can the reparation process can begin [98][50]. In the SVD, due to the chronic hypoxia-hypoperfusion condition [12][13], the passage through M1 towards M2 activation does not occur [98][50]. In SVD, there is a substantial augmentation of M1 activation, together with a heavy reduction of M2 promotion [99,100][51][52]. The brisk oligodendrocyte degeneration, associated with M1 activation, increases calcium currents and induces a severe apoptosis process. The calcium increases, and the severe apoptosis is accompanied by an augmentation of caspase-3 RNA and matrix-metalloprotease 2 (MMP-2) [101][53]. At the beginning of the SVD process, these markers reflect the temptation reparation process induced by a standard M1/M2 passage, as described above. Nevertheless, until the chronic inflammatory condition occurs in SVD ongoing development, there is an alteration of the M1/M2 passage, with a predominant M1 event; therefore, in SVD patients’ cerebrospinal fluid (CSF), there is a constant growth of oligodendrocyte-derived myelin sheath-like myelin lipid sulfatide (ODMSMS) and myelin essential protein (MBP) due to the massive oligodendrocytes death [102,103,104,105][54][55][56][57]. For similar reasons, markers of axonal damage, i.e., neurofilament light chain (NFL), together with CSF α-1 antitrypsin, tissue inhibitor of metalloproteinase-1 (TIMP-1), plasminogen activator inhibitor-1 (PAI-1), and apolipoprotein H (ApoH) have been found to increase very early in the CSF in SVD [106,107,108][58][59][60]. Finally, due to the BBB leakage, ultrastructural studies find that in older animals as well as in those affected by SVD, there are severe alterations of the capillary basement membrane of the deeper arterioles, inside the white matter, filling plasma proteins into vascular bagging and collagen deposition inside PVS, in a phenomenon described as microvascular fibrosis [55,98,109][5][50][61]. Many studies have testified that microvascular fibrosis and BBB splitting have a higher CSF/serum albumin (SA) ratio in patients with SVD [109][61]. Matrix remodeling pathway (TIMP-1 and matrix metalloproteinases) as an expression of endothelium disruption in SVD has been described [109][61].
2. Markers of Oxidative Damages in SVD
Reactive oxygen species (ROS) is an umbrella term for many ordinary derivatives of molecular oxygen, and their accumulation leads to a complex phenomenon called oxidative distress. There are two species, hydrogen peroxide (H2O2) and the superoxide anion radical (O2−), which are key redox signaling agents generated under the control of growth factors and cytokines by more than 40 enzymes, prominently including nicotinamide adenine dinucleotide phosphate (NADPH) oxidases [12][13] and the mitochondrial electron transport chain [126][62]. When mitochondrial cells usually function, the active process of oxidative phosphorylation converts oxygen to superoxide by oxidase enzymes, and superoxide can be transformed by superoxide dismutase (SOD) or to non-radical hydrogen peroxide [126[62][63][64],136,137], for i.example., from glutathione peroxidase (Gpx), or when catalase enzymatically metabolizes hydrogen peroxide to water and oxygen [136][63].
Chronic cerebral conditions of constant hypoxia are the principal inductors of the uncontrolled production of ROS [138,139][65][66].
NADPH oxidase activity and mitochondrial are significantly higher in cerebral arteries when compared with systemic arteries in blood vessels from healthy animals (mouse, rat, pig, and rabbit) [140,141][67][68]. Thus, brain vessels are one of the most prominent productions of ROS, suggesting that there could be fundamental ROS-dependent signaling in cerebral arteries, which might be indispensable for vasoactive regulation properties.
Thus, the accumulation of ROS species, associated with mitochondrial dysfunction, BBB disruption, and chronic inflammatory status are three conditions in SVD and are proportionate to WMH extension. They lead to an altered endothelial further altered activation, which is reflected in a decoupling of the neurovascular coupling system, with significant sub-cortical and cortical signal alteration, with consequent reflex in oligodendrocytes astrocytes and finally to neurons [12,142][13][69]. An active role of flow-dependent responses in rat cerebral arteries has been recently demonstrated in vivo, directly exerted by the NADPH-oxidase reactions [143][70]. Specifically, Nox2-NADPH oxidase dysfunction is related to the propagation of the ischemic brain injury, derived by the occlusion of larger pial arteries; Nox2/NOx2 knock-out mice, in the same condition, show the minor extension of brain injury after an ischemic infarct [144][71].
The induced alterations of mitochondrial DNA by ROS attacks and chronic ischemic conditions are some of the most critical contributors to neuronal aging and degeneration, either considering oxidative damage as a promoter or as a consequence of it [145,146,147][72][73][74].
The decline of mitochondrial functioning has been largely implicated in the aging process and is characterized by a reduced density of mitochondria and reduced mitogenesis [148,149,150,151,152][75][76][77][78][79]. Such changes, which originate as replication errors, accumulate in postmitotic tissues during aging, leading to increased proportions of impaired mitochondria [152][79]. In the aging brain, there has been a sufficient demonstration of impairment of synaptic mitochondria leading to impaired neurotransmission and cognitive failure [149,150,151,152,153,154,155][76][77][78][79][80][81][82]. Precocious forms of small vessel disease, leading to vascular dementia, have been described in specific mitochondrial point mutation [156][83]. Other mitochondrial mutation phenotypes have been described as pure brain involvement, including fluctuating encephalopathy, seizures, dementia, migraine, stroke-like episodes, ataxia, and spasticity [149,153,154,155][76][80][81][82]. Growing attention should be paid to mitochondrial DNA mutations for brain pathologies, in order to gain more robust data on their possible relevance, and their correlation with postmortem neuropathologic features, to advance ourthe understanding [156,157,158,159,160,161][83][84][85][86][87][88].
Oxidative stress potentiates the disorders of the endothelium-dependent NO signaling [162,163][89][90]. Uncoupling endothelial NO synthase (eNOS) (i.for example., in relation with lower levels of tetrahydrobiopterin) switches the production of NO to that of superoxide, causing an overwhelming potentiation of ROS production, accelerating the oxidative stress, lowering the NO anti-inflammatory properties [164[91][92],165], and reducing NO modulation of Rho-kinase activity, inhibiting vascular tone control [166][93]. Rho-kinase, as a counterpart, influences mRNA-stability of eNOS [167][94].
The induction of oxidative stress is one of the most important promoters of pathological angiogenesis, by lipid oxygenation, thickening the blood vessel walls [168,169][95][96]. Moreover, the ApoE4 allele and the AD process seem to be involved in promoting vascular alterations independently of other recognized factors, i.e., age, diabetes, hypertension, and obesity, etcand so on. However, it is supposed to worsen the confluency of WMH, probably somehow linked to ROS augmentation, without any other positive data [170,171,172][97][98][99].