Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sofia Barbosa-Gouveia and Version 2 by Rita Xu.
Mitochondrial functional integrity depends on protein and lipid homeostasis in the mitochondrial membranes and disturbances in their accumulation can cause disease. AGK, a mitochondrial acylglycerol kinase, is not only involved in lipid signaling but is also a component of the TIM22 complex in the inner mitochondrial membrane, which mediates the import of a subset of membrane proteins.
- acylglycerol kinase
- mitochondrial dysfunction
- mitochondrial ATP generation
1. Introduction
Mitochondria are responsible for several vital functions in mammalian cells, including adenosine triphosphate (ATP) production, steroid and stress hormones synthesis, cellular metabolism, and the induction of apoptosis [1][2][3][1,2,3]. Moreover, spatially and functionally regulated specific microdomains known as the mitochondria-associated membranes (MAMs) between the endoplasmic reticulum (ER) and mitochondria are hot spots of Ca2+ transfer, highlighting the mitochondria vital role in cellular Ca2+ homeostasis [4].
Mitochondrial diseases can arise due to defects in nuclear or mitochondrial DNA (nDNA and mtDNA, respectively) genes that encode proteins required for normal mitochondrial structure and/or function [5]. Acylglycerol kinase (AGK) is a mitochondrial membrane protein involved not only in lipid and glycerolipid metabolism but also in mitochondrial protein transport, glycolysis, and thrombocytopoiesis [6][7][8][6,7,8]. AGK can act as a lipid kinase to catalyze the phosphorylation of diacylglycerol (DAG) and monoacylglycerol (MAG) to phosphatidic acid (PA) and lysophosphatidic acid (LPA), respectively. Both PA and LPA can act as signaling molecules and participate in phospholipid synthesis [9][10][9,10], which is essential to maintain mitochondrial structure and function. Moreover, AGK scavenges mitochondrial DAG, which induces reactive oxygen species (ROS) signaling via a pathway mediated by protein kinase D1 [9][11][9,11].
Independently of its lipid kinase activity and its role in lipid metabolism stability, AGK acts as a subunit of the mitochondrial translocase of the inner membrane 22 (TIM22) complex [12][13][12,13], which is responsible for the translocation of transmembrane proteins from the cytoplasm into the mitochondrial interior [14] and their insertion into the mitochondrial inner membrane via the formation of a twin-pore translocase that harnesses membrane potential as the external driving force [14][15][14,15]. The TIM22 complex, which is required to import a subset of metabolite carriers into mitochondria, including ANT1/SLC25A4 and SLC25A24, consists of multiple subunits: Tim22, the core pore-forming subunit; the intermembrane space chaperones Tim9, Tim10, and Tim10b; and AGK and Tim29, which are specific subunits of the complex [14].
Sengers syndrome (OMIM #212350) is a rare autosomal recessive disorder caused by mutations in the AGK gene and characterized by hypertrophic cardiomyopathy, congenital cataracts, and mitochondrial myopathy, including muscle weakness and lactic acidosis after exercise [16]. Two clinical forms have been identified: a severe neonatal form that can cause infantile death due to heart failure consequent to hypertrophic cardiomyopathy, and a benign form with a better prognosis in which surviving patients achieve normal developmental milestones [9][17][9,17]. The pathological mechanism underlying Sengers syndrome remains unclear, although it is thought to involve AGK’s role in lipid metabolism.
2. Molecular Genetics and In Silico Analysis of the AGK Variant
NGS analysis enabled the identification of a homozygous variant, c.518+1G>A, in the AGK gene (NM_018238.4) in this patient. This splicing variant is located in intron 8, and segregation studies performed by Sanger sequencing confirmed that it was inherited from both parents (Figure 1A). The AGK variant was analyzed in silico to determine the degree of evolutionary conservation, predicted pathogenicity, functional consequences, and minor allele frequency (MAF) within the population. In addition, the Human Splicing Finder (HSF) [18] predicted that the splicing variant c.518+1G>A, located in intron 8, disrupts the wild-type splice donor site, likely affecting splicing (Figure 1B). Variants in the canonical acceptor and donor sites have been previously reported to strongly affect conserved sequences [19]. The 5′ splice site (donor site) and 3′ splice site (acceptor site) sequences are recognized by the elements of the spliceosome. Consequently, any variant located in these canonical sequences can alter the interactions between pre-mRNA and proteins involved in intron removal. Variants at the canonical splice sequences usually lead to single exon skipping [20]. The AGK gene encodes a 422-amino acid protein. Sequencing of our patient’s cDNA revealed a decrease in sequence length of 96 bp compared with control cDNA (Figure 1C), an effect likely due to exon 9 (96 bp) skipping during the splicing process.
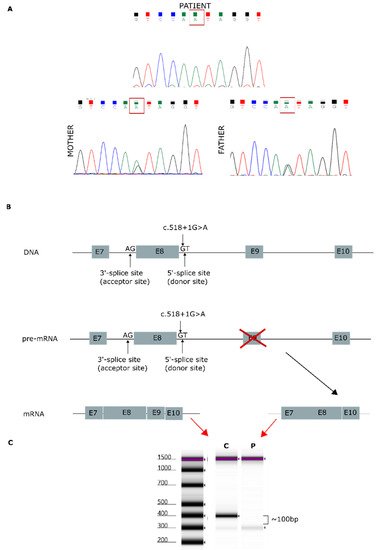
Figure 1. Genetic analysis findings. (A) Genomic DNA sequence chromatograms showing Sanger sequencing results. Both parents carried the splicing variant c.518+1G>A, located in intron 8. (B) AGK gene: predicted effect of exon 9 skipping during mature mRNA splicing. (C) cDNA sequencing of patient [P] and control [C], showing a 96-bp decrease in the length of the AGK cDNA sequence.
3. AGK Protein Modeling
The AGK protein consists of a typical two-domain fold (DGK domain 1 and DGK domain 2) that mediates phosphorylation of monoacylglycerols or diacylglycerols and has an N-terminal α1 helix that anchors to the membrane and a C-terminal key region with an additional membrane anchor helix loop (Figure 2) [6][21][6,21].
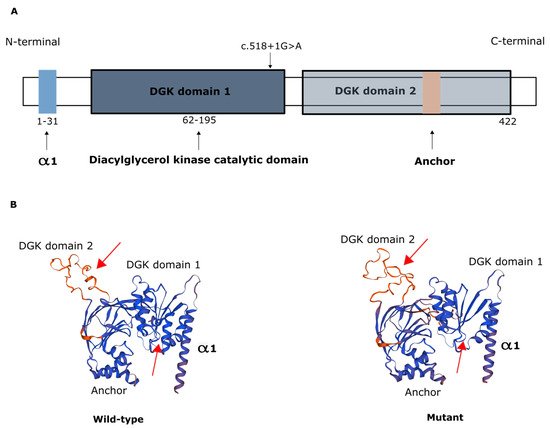
Figure 2. Representation of AGK protein domains. (A) Domain structure of AGK, which contains a two-domain fold (DGK domains 1 and 2) that mediates phosphorylation of monoacylglycerols or diacylglycerols, an N-terminal α1 helix (light blue) that anchors to the membrane, and a C-terminal region with an additional membrane anchor (light brown) helix loop. Schematic depicts the c.518+1G>A variant in the catalytic domain. (B) Protein modeling of wild-type AGK and of the AGK variant c.518+1G>A, showing predicted changes in spatial protein structure (red arrows).
4. Mitochondrial Respiration and OXPHOS Activity
Measurement of the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) revealed decreases in both OCR and the OCR:ECAR ratio in the patient’s fibroblasts, indicating reduced electron flow through the respiratory chain (Figure 3A,B). These findings reflect a general deficiency in the overall function of all mitochondrial respiratory chain complexes in the patient’s fibroblasts.
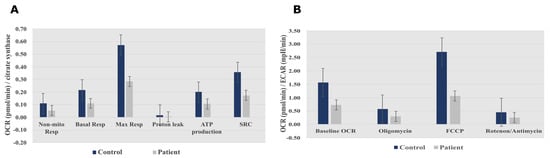
Figure 3. Seahorse XF96 respirometry analysis. (A) Oxygen consumption rate measured before and after the addition of inhibitors. These modulators are electron transport chain inhibitors (oligomycin, FCCP, and a mixture of rotenone and antimycin A), which are serially injected to measure ATP production, Max Resp, Non-Mito Resp, proton leak, SRC, and Basal Resp. (B) The basal energy metabolism of each cell line was assessed by determining OCR:ECAR ratios following sequential injection of the inhibitors. Abbreviations: Basal Resp, basal respiration; ECAR, extracellular acidification rate; FCCP, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; Max Resp, maximal respiration; Non-Mito Resp, non-mitochondrial respiration; OCR, oxygen consumption rate; SRC, spare respiratory capacity. All data were obtained from two independent experiments, expressed as mean value ± deviation standard.
Spectrophotometric analysis of respiratory chain enzyme activity revealed deficiencies in complexes I and V in the patient’s fibroblasts (Table 1).
Table 1. Measurement of enzyme activities for the different OXPHOS complexes in patient and control fibroblasts (reference range determined for healthy population).
| Patient-Activity (mU/U CS) | Control-Activity (mU/U CS) | Reference Range |
|---|
| Complex I | 142 | 320 | 163–599 |
| Complex II | 367 | 582 | 335–888 |
| Complex III | 592 | 628 | 570–1383 |
| Complex IV | 556 | 527 | 288–954 |
| Complex V | 103 | 712 | 193–819 |
Bold font indicates complex for which reduced activity was observed. Abbreviations: CS, citrate synthase.