1. Introduction
Sphingolipids, including sphingomyelin and its metabolites, are structural components of all cell membranes and of the myelin sheath in the nervous system. They were discovered by J.Thudichum in 1874
[1] but it was not until 1997, when the specific, high-affinity G-protein-coupled receptors for the sphingomyelin metabolite sphingosine 1-phosphate (S1P) were identified
[2][3][2,3], that their multiple physiologic roles in the human body started to be recognized. Subsequent molecular and physiologic research studies suggested that the S1P system mediates various intracellular signaling cascades in the CNS, cardiovascular and immune systems
[4]. S1P is a bioactive lipid second messenger that has important signaling functions in cell growth, cell proliferation and angiogenesis. It also regulates biological functions in health and disease
[5][6][5,6]. S1P concentrations are low in intracellular and interstitial fluids and increased within blood and lymph, in the sub-micromolar range, therefore creating a S1P gradient, which is important for regulating physiologic actions such as lymphocyte egress from secondary lymphoid organs. Multiple enzymes are essential for maintaining the S1P gradient between tissues and systemic circulation and a dynamic equilibrium between S1P and sphingosine. S1P is a lipid metabolite of ceramide. Ceramide can be either broken down by ceramidases to sphingosine, or phosphorylated in the Golgi apparatus by ceramide kinase to produce ceramide-1-phosphate (C1P), another sphingolipid metabolite. In contrast with ceramide and sphingosine, which are considered to activate an apoptotic response, S1P is associated with the suppression of apoptosis
[7]. S1P is produced intracellularly through the phosphorylation of sphingosine by sphingosine kinases (SphK1, SphK2), or extracellularly through the hydrolysis of sphingosyl phosphorylcholine by autotoxin. SphK1 is mainly responsible for the cytosolic and extracellular S1P, has trophic functions and can be upregulated in response to proinflammatory cytokines. In contrast, SphK2 can translocate into the nucleus and has the capacity to enhance apoptosis
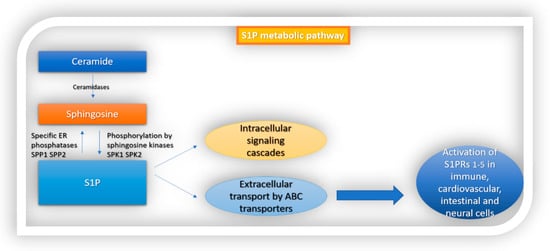
[5]. Following its production, S1P interacts with intracellular targets or is transported extracellularly by ABC transporters, in autocrine or paracrine cell targets, to activate the G-protein-coupled receptor named sphingosine 1-phosphate receptors 1–5 (S1PRs
1–5) (
Figure 1). S1P is converted to sphingosine by S1P-specific ER phosphatases (SPP1 and SPP2) or degraded by S1P lyase (S1PL)
[5][8][9][5,8,9].
Figure 1. S1P metabolic pathway. S1P is formed from ceramide. Ceramidase converts ceramide to sphingosine, which is then phosphorylated by sphingosine kinases 1 and 2 to S1P. Following its production, S1P interacts with intracellular targets or is transported extracellularly by ABC transporters. Finally, it activates sphingosine 1-phosphate receptors 1–5 (S1PRs 1–5) which are ubiquitously expressed in different cell types in the body.
3. S1P Signaling in the Intestine
The gastrointestinal (GI) tract is the largest epithelial barrier that protects the human body from the external environment. Preservation of an intact intestinal mucosal barrier is important for GI functions including digestion of food, absorption of nutrients, expulsion of waste, and protection against hostile bacteria
[32][41]. The proliferation and migration of intestinal epithelial cells (IECs) must be carefully balanced and regulated
[33][42]. This way, the body can preserve the integrity of the intestinal mucosal barrier and manage to maintain the protection of the host from the environment but also the communication between host and its environment. These functions are regulated by a variety of signaling molecules
[34][43]. The intestinal immune system must preserve its immunological homeostasis. The disturbance of the intestinal immune function causes the outbreak of allergic, inflammatory, and infectious diseases
[35][36][44,45]. S1P is a biolipid mediator that controls various cell functions, as described in detail above. In the gastrointestinal system, S1P has important roles in maintaining intestinal epithelial cell barrier structural and functional integrity, regulating IEC proliferation, migration and apoptosis. It also mediates immunological actions like immunoglobulin A production and T cell trafficking
[32][35][36][41,44,45]. In general, S1P is a byproduct of the catalyzation of sphingomyelin and is produced mainly by platelets, erythrocytes, and endothelial cells in mammals. However, in the intestinal tissue the production of S1P is mainly due to epithelial cell activity.
Interestingly, it has been suggested that the levels of dietary sphingomyelin intake and the activity of the enzymes sphingomyelinase and sphingosine kinase may influence the incidence and severity of intestinal inflammatory processes
[35][37][38][39][44,46,47,48]. Furthermore, cholesterol ingested by food antagonizes the intestinal absorption of sphingolipids. Thus, it is implied that modern Western diets, rich in cholesterol, may promote S1P-mediated intestinal immunity dysfunction and the subsequent genesis of inflammatory disorders, through its impact in reducing the availability of S1P precursors
[36][40][45,49]. Recent studies have brought up evidence supporting the role of S1P signaling in the activation of two critical transcription factors, nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3). These transcription factors are known to regulate key inflammatory signaling pathways that promote colon inflammation and carcinogenesis in the intestinal as well as in other human systems
[41][50].
Implication in Intestinal Barrier Homeostasis
S1P is found abundantly in the human intestine. All five S1PRs are expressed in intestinal epithelial cells, as indicated by the findings from the Human Protein Atlas, but the expression level of each one varies, with the highest being that of S1PR2
[42][51]. The role of S1P in intestinal epithelial barrier integrity has been investigated, but still not all its functions have been revealed. Greenspon et.al have demonstrated that, following S1P administration in differentiated rat IECs, intestinal barrier function was enhanced. Treatment with S1P led to an increase in expression of an adherens junction protein, E-cadherin, and also in more efficient redistribution of this protein on the barrier. Increased abundance of S1P, resulting from upregulated expression of SphK1 in IECs, was also related to augmented levels of other barrier proteins including claudin-1 and occludin. These effects were attributed to activation of S1PR1
[43][52]. Chen et al. have shown that S1P also upregulates the expression of c-Myc, cyclin D1, E-cadherin and zona occluden-1 (ZO-1) via activation of S1PR2
[34][43]. These effects result in maintaining the integrity of the intestinal epithelial cell barrier. The above authors later conducted further research experiments, using an S1PR2-knockout mice model, with the administration of Dextran sulfate sodium (DSS) to induce colitis. The pivotal role of S1P-mediated S1PR2 activation in regulating intestinal barrier structural integrity and function was reaffirmed. It was demonstrated that the S1P/S1PR2 axis in IECs prevented intestinal barrier damage by mediating (a) CD4+T-cell activation via the ERK pathway and (b) MHC-II expression. In addition, they found that IFN-γ, secreted by CD4+T cells, augmented DSS-induced damage of the intestinal barrier function. This was done through downregulation of ZO-1 by IFN-γ. On the other hand, S1P was capable of restricting DSS/IFN-γ-induced damage of intestinal mucosa permeability by increasing ZO-1
[44][53]. Additionally, S1P has been found to promote intestinal epithelial cell proliferation and migration, in a dose-dependent manner. This is achieved through S1P-mediated activation of ERK1/2 via S1PR2
[34][43]. Furthermore, S1P-mediated activation of S1PR2 leads to increased expression of down-regulated in adenoma (DRA), the major Cl-/HCO3-exchanger that regulates NaCl absorption in the intestine of mammals
[45][54]. MEK/ERK activity controls JNK activation via regulating MAPK phosphatase (MKP-1), which prevents cell apoptosis. S1P enhances the ERK1/2 and Akt signaling pathways thus preventing intestinal epithelial cells’ apoptosis
[46][47][55,56] (
Table 1).
4. S1P Signaling in the CNS
S1P is widely present in the CNS, where it is released by cerebral sphingosines and acts on S1P receptors. All S1PRs except S1PR4 are expressed in the CNS by neuronal and glial cell populations and also in the cerebral vascular system, to varying degrees. Their activation by S1P mediates numerous physiologic processes involved in neuronal plasticity including myelination, neurogenesis and neuroprotection
[48][57]. Evidence from animal models and studies in vitro suggest that S1P regulates multiple physiologic functions in the CNS and that the expression of S1PRs is dynamic, influenced by temporal and spatial factors and stimuli of the cell environment
[4][49][50][4,58,59].
In neurons, S1P signaling has an active role in neural progenitor migration, synaptic activity (=neurotransmission), differentiation, process extension, calcium signaling and survival
[51][60]. S1P stimulates neurogenesis more effectively than fibroblast growth factor (FGF)
[52][61]. S1P binding to S1PR1, S1PR2 and S1PR5 participates in the regulation of growth cone formation, neurite extension and retraction. In vitro experiments of stimulation of neurons with nerve growth factor (NGF) showed the resulting enhancement of neurite extension which is mediated by S1PR1. Opposingly, the activation of S1PR2 and S1PR5 inhibits neurite extension
[53][62]. Several researchers have addressed the role of S1P-mediated circuits in neural progenitor stem cells (NPSCs). The effect of S1P on NPSCs is mediated by increased laminin expression and extracellular matrix (ECM) interactions with progenitor integrins
[54][63]. NPSCs that migrate out of the embryoid body stem cells (ESCs) are considered to upregulate S1PR1
[55][64]. However, there are important variations between studies concerning the description of S1PR expression in NPSCs
[52][61]. S1PR1 also affects plasma membrane excitability and neurotransmitter release, thus promoting synaptic transmission, as demonstrated in studies with rat hippocampus models
[56][57][65,66]. Control of neural activity has been attributed to S1PR2 signaling
[58][67], as models of S1PR2-knockout mice have resulted in excess neural excitability and the occurrence of seizures at 3–7 weeks of age
[51][59][60,68]. The regulation of synaptic activity, neurogenesis and cell survival is such an important factor that these receptors have attracted interest as potential drug targets in memory disorders and neurodegenerative diseases.
Oligodendrocytes and oligodendrocyte precursor cells (OPCs) widely express S1PRs, and the level of each S1PR’s expression depends on the cells’ stage of development and on the myelinating state
[49][60][61][58,69,70]. In mature oligodendrocytes, S1PR5 expression is highest and is thought to promote cell survival via the S1PR5-mediated Akt signaling pathway and inhibit OPC migration via the S1PR5-mediated Rho GTPase/Rho kinase pathway
[62][71], whereas it is assumed that early stages of differentiation are mainly mediated by S1PR1 signaling
[50][59]. Indeed, various authors suggest that higher expression level and activation of S1PR1 induce the differentiation of oligodendrocytes along with the survival of oligodendrocyte progenitors
[52][63][61,72]. Regarding the role of S1P in oligodendrocyte morphology, there is an interesting dual action. Via the modulation of S1PR5, it enhances process retraction
[64][73], while via S1PR1, it promotes process extension, indicating reciprocal effects on cytoskeletal elements. S1PR5 is also considered to have a pivotal role in myelination, although data from S1PR5-null mice showed that myelination was normally developed despite the lack of S1PR5 expression
[64][73]. On the other hand, the targeted deletion of S1PR1 in oligodendrocyte lineage cells led to abnormal formation of myelin and increased susceptibility to cuprizone-induced demyelination
[65][74]. This finding suggests an important role for S1PR1 in normal myelination of the brain. S1P actions on oligodendrocytes and OPCs are also influenced by the action of neurotrophic factors and the lysophosphatidic acid receptors expressed on these cells, such as neurotrophin-3 and platelet-derived growth factor. It seems very probable that the process of myelination is coordinated with the contribution of all the above cellular structures
[51][66][60,75]. Altogether, interactions between S1P and S1PRs in oligodendrocytes and OPCs control cell survival, migration and differentiation, maintenance of cell morphology, myelination and remyelination.
S1P signaling mediates various functions in astrocytes, including differentiation
[52][67][61,76], proliferation, migration, gap junction communication, growth factor production and astrogliosis
[50][51][59,60]. Astrocytes mainly express S1PR1 and S1PR3, with scarce expression of S1PR2. S1PR5 expression by astrocytes was detected during in vitro studies, when exposed to growth factors
[68][77]. Astrocyte proliferation is stimulated by S1P via activation of the extracellular-signal-regulated kinase (ERK) pathway
[69][78]. Observations of activated astrocytes in response to pathogens suggest that they overexpress S1PR1 and S1PR3, pointing to a role of these receptors in astrogliosis
[70][79]. Studies in S1PR-knockout mice also suggest that astrogliosis requires the activation of S1PR1 and S1PR3
[71][80]. Some research groups support that S1PR3 is overexpressed in astrocytes under proinflammatory conditions
[52][67][61,76]. Interestingly, S1P antagonism by fingolimod reduces the production of the inflammatory chemokines CXCL5/LIX, C-X-C motif chemokine 10 (CXCL10) and monocyte chemoattractant protein-1 (MCP-1) in astrocytes and microglial cultures
[72][81]. Therefore, S1P signaling in astrocytes seems to mediate both proinflammatory and anti-inflammatory effects.
S1PR1, S1PR2 and S1PR3 are also expressed by microglia. Microglia exist in a nonactivated state and switch to an activated state when stimulated by proinflammatory cytokines. The expression of S1PRs in microglia cells depends on their activation state
[73][82]. Extracellular S1P is a powerful chemoattractant for microglial cells in the brain
[52][61]. S1P levels have been shown to increase at sites of brain damage, where microglia and neural progenitors accumulate. S1PR1 and S1PR3 are downregulated in activated microglia, and S1PR2 is upregulated. In models of experimental autoimmune encephalomyelitis (EAE), S1PR1 deletion was correlated with reduced activation of microglia cells
[72][81]. (
Table 1)
Implication in BBB Homeostasis
Plasma S1P-knockout mice display increased morbidity due to increased vascular leak and anaphylaxis following the administration of platelet-activating factor or histamine
[74][83]. Likewise, pharmacologic blockage of S1PR1 or ApoM deficiency leads to compromised vascular integrity and excess inflammation
[75][84]. Brain endothelial SP1 signaling also supports the BBB integrity by arranging the localization of tight junction proteins
[76][85]. Maintenance of vascular integrity requires proper rearrangements of the cytoskeleton as well as the assembly of adherens junctions in endothelial cells. S1P promotes the structural integrity of the actin cytoskeleton, the accumulation of VE-cadherin and α-, β- and γ-catenin at the sites of adjacent cell contact, and adherens junction assembly
[77][86]. The activation of small G proteins Rac and Rho downstream of the S1PR1 and S1PR3 signaling pathways are involved in the processes
[77][78][86,87]. S1P−S1PR1 signaling is considered to have a pivotal function in regulating the inflammatory status of vascular endothelial cells. Giovani et al. have shown that endothelial S1PR1 abundance was enhanced in regions of vascular inflammation. Additionally, proinflammatory adhesion proteins such as VCAM-1 and ICAM-1 were upregulated in the descending aorta of mice with endothelial cell-specific deletion of S1PR1 and suppressed in mice with endothelial cell-specific overexpression of S1PR1
[79][12]. These observations indicate that proper S1PR1 localization and signaling are important to maintain vascular homeostasis, and that the impairment of S1PR1 signaling because of receptor internalization predisposes endothelial cells to an inflammatory phenotype
[14][11].
5. Implication for the Role of S1P in Multiple Sclerosis
The ubiquitous presence of S1P and its receptors in the CNS implies that the S1P−S1PR signaling system may be targeted for therapeutic purposes in neurological disorders like MS
[80][88]. MS is a T cell-mediated autoimmune disease of the CNS and its etiology is believed to constitute an interaction between genetic predisposition and environmental factors. MS is classified as follows, according to the clinical course of the disease: relapsing–remitting MS, primary progressive MS, and secondary progressive MS
[81][89].
The activation and clonal expansion of CNS-directed, autoreactive CD8+ T cells, differentiated CD4+ TH1 and TH17 cells, B cells and innate immune cells in the periphery are considered to be the first step in MS pathogenesis
[82][90]. Following this, T cells cross the BBB, with the interference of integrins and, upon entrance, they are reactivated by epitopes on myelin and induce inflammation with the release of cytokines such as tumor necrosis factor alpha (TNFα) and IFNγ. These events lead to increased BBB permeability, demyelination and axonal degeneration
[81][83][89,91]. After their activation by TNFα and IFNγ, CNS resident microglia and infiltrating macrophages are transferred to the site of inflammation, also contributing to destruction of myelin and prolonging neurodegeneration
[84][92]. The B lymphocytes’ role in the pathogenesis of the disease has attracted vivid interest and thorough research in the last years, and therapeutic agents targeting these cells have shown positive results in limiting disease progress
[85][93]. B cells contribute to MS pathophysiology through multiple mechanisms such as antigen presentation to T cells, transport of antigens from tissues to secondary lymphoid organs and the release of proinflammatory or anti-inflammatory cytokines. Pathogenetic auto-antibodies have been identified in a subgroup of MS patients
[86][94].
Following the acute inflammatory attack, the patient exhibits functional recovery that is attributed to the remyelination of the axons and brain plasticity. However, axonal damage, which is secondary to demyelination and neurotoxic factors released from activated microglia, is not reversible. Accumulating axonal damage leads to neuronal degeneration and brain atrophy, which is the main contributor of progressive irreversible neurological disability
[48][57]. Another residue of the acute inflammatory event is the subsequent chronic inflammation which is restricted within the CNS. This is due to the persistence of activated immune cells in the CNS, despite the absence of infiltrating lymphocytes from the periphery. This chronic inflammatory process affects the whole brain parenchyma, even at sites far from the underlying focal demyelinating lesions. Diffuse chronic CNS inflammation is considered more prevalent in patients with progressive forms of MS
[48][87][57,95]. Inflammation in MS is thus differentiated both in the space axis (peripheral vs. local CNS inflammation) and in the time axis (acute vs. chronic inflammation). Both inflammatory and neurodegenerative components of the disease are thought to be present from the early stages, and proceed in parallel, but are responsible for different aspects of the clinical phenotype.