Air pollutants include toxic particles and gases emitted in large quantities from many different combustible materials. They also include particulate matter (PM) and ozone, and biological contaminants, such as viruses and bacteria, which can penetrate the human airway and reach the bloodstream, triggering airway inflammation, dysfunction, and fibrosis.
- air pollutants
- airway disease
- asthma
- COPD
1. Introduction
2. The Effects of Air Pollutants on Asthma and COPD, Upper Airway Disease
Air pollution is one of the most important environmental factors affecting public health, due to its effects on the respiratory system [40][5]. Air pollution can interfere with defense mechanisms in the lung, weaken the body’s immune response [41][6], and trigger oxidative stress and inflammation [42,43][7][8]. Air pollution is defined as the presence of aerial substances that are harmful to humans, and is associated with a higher risk of premature death due to cardiovascular diseases (e.g., ischemic heart disease and stroke), asthma, COPD, lower respiratory tract infections, and lung cancer [31,40][9][5]. Ozone has a strong smell and irritates the respiratory system, resulting in swelling of the throat, discomfort in the chest, coughing, sputum production, and even emphysema with long-term exposure [44][10]. Nitric oxide may generate photochemical smog and has acute toxic effects on the human lungs. The short- and long-term effects of PM10, PM2.5, and SO2 on lung function, disease morbidity, and mortality have been described in detail [23,40,45][11][5][12]. Air pollution is associated with various respiratory and non-respiratory diseases including asthma, COPD, pneumonia, lung malignancies, heart disease, stroke, dementia, and diabetes [46,47,48,49,50][13][14][15][16][17].
The long-term effects of air pollution on asthma were summarized in an American Thoracic Society workshop report, which indicated that long-term exposure to air pollution was a cause of childhood asthma. However, there was insufficient evidence to draw a similar conclusion regarding adult asthma [65][18]. Many studies have described correlations between short-term exposure to outdoor air pollutants and various aspects of asthma, including symptom control [66][19], lung function [67][20], medication dose [68[21][22],69], outpatient visits [70[23][24],71], asthma exacerbations [72,73][25][26], emergency room visits [74][27], hospitalizations [75,76][28][29], length of hospital stay [77][30], and mortality rate [78][31] (Figure 1).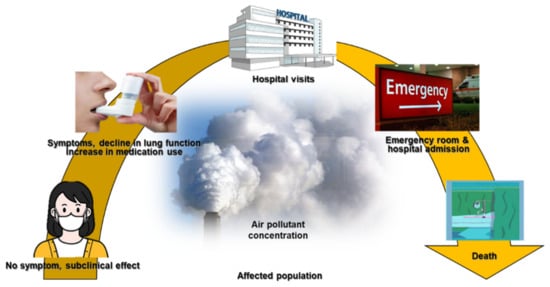
The most common cause of COPD is smoking; some cases are also due to air pollution and genetics [82][32]. Poorly ventilated cooking fires, often using coal or biomass fuels such as wood, lead to indoor air pollution and are a common cause of COPD in developing countries [83][33]. Such fires are used by nearly 3 billion people for cooking and heating, and adverse effects on health are more frequent in women due to their higher levels of exposure [84,85][34][35]. These fires are the main source of energy in 80% of all homes in India, China, and sub-Saharan Africa [85][35].
Several epidemiological studies have shown that air pollutants exacerbate airway diseases such as AR, asthma, bronchitis, and COPD. Pollutants such as TRAPs also have negative effects on other upper airway diseases such as AR and non-AR, sinusitis, and otitis media [87][36]. Increasing evidence suggests that PM, photochemical pollutants, and ozone are also linked to the development of upper airway diseases [87][36]. Young children and individuals who are obese are particularly susceptible to these conditions [87][36]. ROS, apoptosis, and inflammation are all involved in the pathophysiological etiology of upper airway diseases [87][36]. Although the data conflict, and controlled prospective studies are needed to determine the relevant mechanisms and risk factors, traffic fumes and tobacco smoke are major factors exacerbating upper airway diseases [87][36].
3. Airway Toxicity Mechanisms Related to Air Pollutants
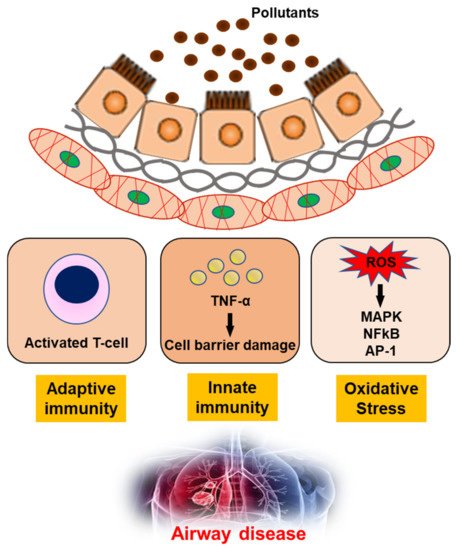
4. Conclusions
References
- McCormack, M.C.; Breysse, P.N.; Hansel, N.N.; Matsui, E.C.; Tonorezos, E.S.; Curtin-Brosnan, J.; Williams, D.L.; Buckley, T.J.; Eggleston, P.A.; Diette, G.B. Common household activities are associated with elevated particulate matter concentrations in bedrooms of inner-city Baltimore preschool children. Environ. Res. 2008, 106, 148–155.
- Di Filippo, P.; Pomata, D.; Riccardi, C.; Buiarelli, F.; Castellani, F.; Calitri, G.; Simonetti, G.; Sonego, E.; Bruni, E.; Uccelletti, D. Concentrations of bacteria and bacterial and fungal spores calculated from chemical tracers associated with size-segregated aerosol in a composting plant. Air Qual. Atmos. Health 2020, 13, 469–476.
- Douglas, P.; Robertson, S.; Gay, R.; Hansell, A.L.; Gant, T.W. A systematic review of the public health risks of bioaerosols from intensive farming. Int. J. Hyg. Environ. Health 2018, 221, 134–173.
- Humbal, C.; Gautam, S.; Trivedi, U. A review on recent progress in observations, and health effects of bioaerosols. Environ. Int. 2018, 18, 189–193.
- Dockery, D.W.; Pope, C.A. Acute respiratory effects of particulate air pollution. Annu. Rev. Public Health 1994, 15, 107–132.
- Olivieri, D.; Scoditti, E. Impact of environmental factors on lung defences. Eur. Respir. Rev. 2005, 14, 51–56.
- Bai, L.; Su, X.; Zhao, D.; Zhang, Y.; Cheng, Q.; Zhang, H.; Wang, S.; Xie, M.; Su, H. Exposure to traffic-related air pollution and acute bronchitis in children: Season and age as modifiers. J. Epidemiol. Community Health 2018, 72, 426–433.
- Karimi, A.; Shirmardi, M.; Hadei, M.; Birgani, Y.T.; Neisi, A.; Takdastan, A.; Goudarzi, G. Concentrations and health effects of short- and long-term exposure to PM2.5, NO2, and O3 in ambient air of Ahvaz city, Iran (2014–2017). Ecotoxicol. Environ. Saf. 2019, 180, 542–548.
- Park, J.; Kim, H.J.; Lee, C.H.; Lee, C.H.; Lee, H.W. Impact of long-term exposure to ambient air pollution on the incidence of chronic obstructive pulmonary disease: A systematic review and meta-analysis. Environ. Res. 2021, 194, 110703.
- Lin, Y.K.; Chang, S.C.; Lin, C.; Chen, Y.C.; Wang, Y.C. Comparing ozone metrics on associations with outpatient visits for respiratory diseases in Taipei Metropolitan area. Environ. Pollut. 2013, 177, 177–184.
- Devlin, R.B.; McDonnell, W.F.; Mann, R.; Becker, S.; House, D.E.; Schreinemachers, D.; Koren, H.S. Exposure of humans to ambient levels of ozone for 6.6 h causes cellular and biochemical changes in the lung. Am. J. Respir. Cell Mol. Biol. 1991, 4, 72–81.
- Stojić, S.S.; Stanišić, N.; Stojić, A.; Šoštarić, A. Single and combined effects of air pollutants on circulatory and respiratory system-related mortality in Belgrade, Serbia. J. Toxicol. Environ. Health A 2016, 79, 17–27.
- Yang, X.Y.; Wen, B.; Han, F.; Wang, C.; Zhang, S.P.; Wang, J.; Xu, D.Q.; Wang, Q. Acute Effects of Individual Exposure to Fine Particulate Matter on Pulmonary Function in Schoolchildren. Biomed. Environ. Sci. 2020, 33, 647–659.
- Genc, S.; Zadeoglulari, Z.; Fuss, S.H.; Genc, K. The adverse effects of air pollution on the nervous system. J. Toxicol. 2012, 2012, 782462.
- Kelly, F.J.; Fussell, J.C. Air pollution and airway disease. Clin. Exp. Allergy 2011, 41, 1059–1071.
- Faustini, A.; Stafoggia, M.; Colais, P.; Berti, G.; Bisanti, L.; Cadum, E.; Cernigliaro, A.; Mallone, S.; Scarnato, C.; Forastiere, F. Air pollution and multiple acute respiratory outcomes. Eur. Respir. J. 2013, 42, 304–313.
- Watson, K.E. Air pollution and heart disease. Rev. Cardiovasc. Med. 2006, 7, 44.
- Thurston, G.D.; Balmes, J.R.; Garcia, E.; Gilliland, F.D.; Rice, M.B.; Schikowski, T.; Van Winkle, L.S.; Annesi-Maesano, I.; Burchard, E.G.; Carlsten, C.; et al. Outdoor Air Pollution and New-Onset Airway Disease. An Official American Thoracic Society Workshop Report. Ann. Am. Thorac. Soc. 2020, 17, 387–398.
- Li, Z.; Xu, X.; Thompson, L.A.; Gross, H.E.; Shenkman, E.A.; DeWalt, D.A.; Huang, I.-C. Longitudinal Effect of Ambient Air Pollution and Pollen Exposure on Asthma Control: The Patient-Reported Outcomes Measurement Information System (PROMIS) Pediatric Asthma Study. Acad. Pediatr. 2019, 19, 615–623.
- Mentz, G.; Robins, T.G.; Batterman, S.; Naidoo, R.N. Effect modifiers of lung function and daily air pollutant variability in a panel of schoolchildren. Thorax 2019, 74, 1055–1062.
- Williams, A.M.; Phaneuf, D.J.; Barrett, M.A.; Su, J.G. Short-term impact of PM2.5 on contemporaneous asthma medication use: Behavior and the value of pollution reductions. Proc. Natl. Acad. Sci. USA 2019, 116, 5246–5253.
- Pepper, J.R.; Barrett, M.A.; Su, J.G.; Merchant, R.; Henderson, K.; Van Sickle, D.; Balmes, J.R. Geospatial-temporal analysis of the impact of ozone on asthma rescue inhaler use. Environ. Int. 2020, 136, 105331.
- Kowalska, M.; Skrzypek, M.; Kowalski, M.; Cyrys, J. Effect of NOx and NO2 Concentration Increase in Ambient Air to Daily Bronchitis and Asthma Exacerbation, Silesian Voivodeship in Poland. Int. J. Environ. Res. Public Health 2020, 17, 754.
- Lu, P.; Zhang, Y.; Lin, J.; Xia, G.; Zhang, W.; Knibbs, L.D.; Morgan, G.G.; Jalaludin, B.; Marks, G.; Abramson, M.; et al. Multi-city study on air pollution and hospital outpatient visits for asthma in China. Environ. Pollut. 2020, 257, 113638.
- Zuo, B.; Liu, C.; Chen, R.; Kan, H.; Sun, J.; Zhao, J.; Wang, C.; Sun, Q.; Bai, H. Associations between short-term exposure to fine particulate matter and acute exacerbation of asthma in Yancheng, China. Chemosphere 2019, 237, 124497.
- Garcia, E.; Berhane, K.T.; Islam, T.; McConnell, R.; Urman, R.; Chen, Z.; Gilliland, F.D. Association of Changes in Air Quality with Incident Asthma in Children in California, 1993–2014. JAMA 2019, 321, 1906–1915.
- Chi, R.; Li, H.; Wang, Q.; Zhai, Q.; Wang, D.; Wu, M.; Liu, Q.; Wu, S.; Ma, Q.; Deng, F.; et al. Association of emergency room visits for respiratory diseases with sources of ambient PM2.5. J. Environ. Sci. 2019, 86, 154–163.
- Alcala, E.; Brown, P.; Capitman, J.A.; Gonzalez, M.; Cisneros, R. Cumulative Impact of Environmental Pollution and Population Vulnerability on Pediatric Asthma Hospitalizations: A Multilevel Analysis of CalEnviroScreen. Int. J. Environ. Res. Public Health 2019, 16, 2683.
- Marques Mejías, M.A.; Tomás Pérez, M.; Hernández, I.; López, I.; Quirce, S. Asthma Exacerbations in the Pediatric Emergency Department at a Tertiary Hospital: Association with Environmental Factors. J. Investig. Allergol. Clin. Immunol. 2019, 29, 365–370.
- Baek, J.; Kash, B.A.; Xu, X.; Benden, M.; Roberts, J.; Carrillo, G. Association between Ambient Air Pollution and Hospital Length of Stay among Children with Asthma in South Texas. Int. J. Environ. Res. Public Health 2020, 17, 3812.
- Liu, Y.; Pan, J.; Zhang, H.; Shi, C.; Li, G.; Peng, Z.; Ma, J.; Zhou, Y.; Zhang, L. Short-Term Exposure to Ambient Air Pollution and Asthma Mortality. Am. J. Respir. Crit. Care Med. 2019, 200, 24–32.
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351.
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365.
- Kennedy, S.M.; Chambers, R.; Du, W.; Dimich-Ward, H. Environmental and occupational exposures: Do they affect chronic obstructive pulmonary disease differently in women and men? Proc. Am. Thorac. Soc. 2007, 4, 692–694.
- Pirozzi, C.; Scholand, M.B. Smoking cessation and environmental hygiene. Med. Clin. N. Am. 2012, 96, 849–867.
- Jang, A.S.; Jun, Y.J.; Park, M.K. Effects of air pollutants on upper airway disease. Curr. Opin. Allergy Clin. Immunol. 2016, 16, 13–17.
- Jang, A.S.; Choi, I.S.; Koh, Y.I.; Park, C.S.; Lee, J.S. The relationship between alveolar epithelial proliferation and airway obstruction after ozone exposure. Allergy 2002, 57, 737–740.
- Schraufnagel, D.E.; Balmes, J.R.; Cowl, C.T.; De Matteis, S.; Jung, S.H.; Mortimer, K. Air Pollution and Noncommunicable Diseases A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 1: The Damaging Effects of Air Pollution. Chest 2019, 155, 409–416.
- Guarnieri, M.; Balmes, J.R. Outdoor air pollution and asthma. Lancet 2014, 383, 1581–1592.
- Sly, P.D.; Cormier, S.A.; Lomnicki, S.; Harding, J.N.; Grimwood, K. Environmentally Persistent Free Radicals Linking Air Pollution and Poor Respiratory Health? Am. J. Respir. Crit Care Med. 2019, 200, 1062–1063.
- Thurston, G.D.; Kipen, H.; Annesi-Maesano, I.; Balmes, J.; Brook, R.D.; Cromar, K. A joint ERS/ATS policy statement what constitutes an adverse health effect of air pollution? An analytical framework. Eur. Respir. J. 2017, 49, 1600419.
- Nel, A.; Xia, T.; Madler, L.; Li, N. Toxic potential of materials at the nanolevel. Science 2006, 311, 622–627.
- Al-Hegelan, M.; Robert, M.T.; Christian, C.; John, W.H. Ambient ozone and pulmonary innate immunity. Immunol. Res. 2011, 49, 173–191.
- Jang, A.S.; Choi, I.S.; Yang, S.Y.; Kim, Y.G.; Lee, J.H.; Park, S.W.; Park, C.S. Antioxidant responsiveness in BALB/c mice exposed to ozone. Respiration 2005, 72, 79–84.
- Jang, A.S.; Choi, I.S.; Takizawa, H.; Rhim, T.; Lee, J.H.; Park, S.W.; Park, C.S. Additive effect of diesel exhaust particulates and ozone on airway hyperresponsiveness and inflammation in a mouse model of asthma. J. Korean Med. Sci. 2005, 20, 759–763.
- Jang, A.S.; Choi, I.S.; Lee, J.U.; Park, S.W.; Lee, J.H.; Park, C.S. The NOS isoforms play different roles in airway inflammation after ozone exposure. Respir. Res. 2004, 5, 5–11.
- Jang, A.S.; Choi, I.S.; Kim, S.W.; Song, B.C.; Yeum, C.H.; Jung, J.Y. Airway obstruction after acute ozone exposure in BALB/c mice using barometric plethysmography. Korean J. Intern. Med. 2003, 18, 1–5.
- Leikauf, G.D.; Kim, S.H.; Jang, A.S. Mechanisms of ultrafine particle-induced respiratory health effects. Exp. Mol. Med. 2020, 52, 329–337.
- Kim, B.G.; Lee, P.H.; Lee, S.H.; Park, M.K.; Jang, A.S. Effect of TiO2 Nanoparticles on Inflammasome-Mediated Airway Inflammation and Responsiveness. Allergy Asthma Immunol. Res. 2017, 9, 257–264.
- Cha, M.H.; Rhim, T.; Kim, K.H.; Jang, A.S.; Paik, Y.K.; Park, C.S. Proteomic identification of macrophage migration-inhibitory factor upon exposure to TiO2 particles. Mol. Cell Proteom. 2007, 6, 56–63.
- Kim, B.G.; Lee, P.H.; Lee, S.H.; Kim, Y.E.; Shin, M.Y.; Kang, Y.; Bae, S.H.; Kim, M.J.; Rhim, T.; Park, C.S.; et al. Long-Term Effects of Diesel Exhaust Particles on Airway Inflammation and Remodeling in a Mouse Model. Allergy Asthma Immunol. Res. 2016, 8, 246–256.
- Kim, B.G.; Park, M.K.; Lee, P.H.; Lee, S.H.; Hong, J.; Aung, M.M.M.; Moe, K.T.; Han, N.Y.; Jang, A.S. Effects of nanoparticles on neuroinflammation in a mouse model of asthma. Respir. Physiol. Neurobiol. 2020, 271, 103292.
- Kim, T.H.; Jang, A.S.; Lee, T.H.; Kim, Y.J.; Lee, E.J.; Kim, J.M.; Park, J.S.; Park, S.W.; Park, C.S. Particle stimulation dephosphorylates glutathione S-transferase π1 of epithelial cells. Toxicology 2011, 284, 12–18.
- Song, H.M.; Jang, A.S.; Ahn, M.H.; Takizawa, H.; Lee, S.H.; Kwon, J.H.; Lee, Y.M.; Rhim, T.Y.; Park, C.S. Ym1 and Ym2 expression in a mouse model exposed to diesel exhaust particles. Environ. Toxicol. 2008, 23, 110–116.
- Kast, J.I.; Wanke, K.; Soyka, M.B.; Wawrzyniak, P.; Akdis, D.; Kingo, K.; Rebane, A.; Akdis, C.A. The broad spectrum of interepithelial junctions in skin and lung. J. Allergy Clin. Immunol. 2012, 130, 544–547.e4.
- Schulzke, J.D.; Gunzel, D.; John, L.J.; Fromm, M. Perspectives on tight junction research. Ann. N. Y. Acad. Sci. 2012, 1257, 1–19.
- Holgate, S.T. Epithelium dysfunction in asthma. J. Allergy Clin. Immunol. 2007, 120, 1233–1244.
- Sweerus, K.; Lachowicz-Scroggins, M.; Gordon, E.; LaFemina, M.; Huang, X.; Parikh, M.; Kanegai, C.; Fahy, J.V.; Frank, J.A. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J. Allergy Clin. Immunol. 2017, 139, 72–81.e1.
- Lee, Y.G.; Lee, S.H.; Hong, J.; Lee, P.H.; Jang, A.S. Titanium dioxide particles modulate epithelial barrier protein, Claudin 7 in asthma. Mol. Immunol. 2021, 132, 209–216.
- Inoue, H.; Akimoto, K.; Homma, T.; Tanaka, A.; Sagara, H. Airway Epithelial Dysfunction in Asthma: Relevant to Epidermal Growth Factor Receptors and Airway Epithelial Cells. J. Clin. Med. 2020, 9, 3698.
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692.
- Grainge, C.L.; Davies, D.E. Epithelial Injury and Repair in Airways Diseases. Chest 2013, 144, 1906–1912.
- Erzurum, S.C. New insights in oxidant biology in asthma. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 1), S35–S39.
- Schroer, K.T.; Gibson, A.M.; Sivaprasad, U.; Bass, S.A.; Ericksen, M.B.; Wills-Karp, M.; Lecras, T.; Fitzpatrick, A.M.; Brown, L.A.; Stringer, K.F.; et al. Downregulation of glutathione S-transferase pi in asthma contributes to enhanced oxidative stress. J. Allergy Clin. Immunol. 2011, 128, 539–548.
- Bucchieri, F.; Puddicombe, S.M.; Lordan, J.L.; Richter, A.; Buchanan, D.; Wilson, S.J.; Ward, J.; Zummo, G.; Howarth, P.H.; Djukanović, R.; et al. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am. J. Respir. Cell Mol. Biol. 2002, 27, 179–185.
- Durack, J.; Lynch, S.V.; Nariya, S.; Bhakta, N.R.; Beigelman, A.; Castro, M.; Dyer, A.M.; Israel, E.; Kraft, M.; Martin, R.J.; et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J. Allergy Clin. Immunol. 2017, 140, 63–75.
- Huang, Y.J.; Nariya, S.; Harris, J.M.; Lynch, S.V.; Choy, D.F.; Arron, J.R.; Boushey, H. The airway microbiome in patients with severe asthma: Associations with disease features and severity. J. Allergy Clin. Immunol. 2015, 136, 874–884.
- Goleva, E.; Jackson, L.P.; Harris, J.K.; Robertson, C.E.; Sutherland, E.R.; Hall, C.F.; Good, J.T., Jr.; Gelfand, E.W.; Martin, R.J.; Leung, D.Y. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1193–1201.
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L.; et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5, e857810.
- Loxham, M.; Davies, D.E. Phenotypic and genetic aspects of epithelial barrier function in asthmatic patients. J. Allergy Clin. Immunol. 2017, 139, 1736–1751.
- Holgate, S.T. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011, 242, 205–219.
- Lee, P.H.; Kim, B.G.; Park, M.K.; Hong, J.; Lee, Y.G.; Jang, A.S. The Impact of Diesel Exhaust Particles on Tight Junctional Proteins on Nose and Lung in a Mouse Model. Allergy Asthma Immunol. Res. 2021, 13, 350–352.
- Lee, P.H.; Hong, J.; Jang, A.S. N-acetylcysteine decreases airway inflammation and responsiveness in asthma by modulating claudin 18 expression. Korean J. Intern. Med. 2020, 35, 1229–1237.
- Kim, B.G.; Lee, P.H.; Lee, S.H.; Park, C.S.; Jang, A.S. Impact of ozone on claudins and tight junctions in the lungs. Environ. Toxicol. 2018, 33, 798–806.
- Moon, K.Y.; Lee, P.H.; Kim, B.G.; Park, C.S.; Leikauf, G.D.; Jang, A.S. Claudin 5 in a murine model of allergic asthma: Its implication and response to steroid treatment. J. Allergy Clin. Immunol. 2015, 136, 1694–1696.
- Kim, B.G.; Lee, P.H.; Lee, S.H.; Baek, A.R.; Park, J.S.; Lee, J.; Park, S.W.; Kim, D.J.; Park, C.S.; Jang, A.S. Impact of the Endothelial Tight Junction Protein Claudin-5 on Clinical Profiles of Patients With COPD. Allergy Asthma Immunol. Res. 2018, 10, 533–542.
- Smallcombe, C.C.; Harford, T.J.; Linfield, D.T.; Lechuga, S.; Bokun, V.; Piedimonte, G.; Rezaee, F. Titanium dioxide nanoparticles exaggerate respiratory syncytial virus-induced airway epithelial barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L481–L496.
- Tatsuta, M.; Kan-o, K.; Ishii, Y.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Ogawa, A.; Fujita, A.; Nakanishi, Y.; Matsumoto, K. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: Protective role of cathelicidin LL-37. Respir. Res. 2019, 20, 1–14.
- Jang, A.S.; Concel, V.J.; Bein, K.; Brant, K.A.; Liu, S.; Pope-Varsalona, H.; Dopico, R.A., Jr.; Di, Y.P.; Knoell, D.L.; Barchowsky, A.; et al. Endothelial dysfunction and claudin 5 regulation during acrolein-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2011, 44, 483–490.
- Kim, B.G.; Lee, P.H.; Lee, S.H.; Hong, J.; Jang, A.S. Claudins, VEGF, Nrf2, Keap1, and Nonspecific Airway Hyper-Reactivity Are Increased in Mice Co-Exposed to Allergen and Acrolein. Chem. Res. Toxicol. 2019, 32, 139–145.
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169.
- Uh, S.T.; Koo, S.M.; Kim, Y.; Kim, K.; Park, S.; Jang, A.S.; Kim, D.; Kim, Y.H.; Park, C.S. The activation of NLRP3-inflammsome by stimulation of diesel exhaust particles in lung tissues from emphysema model and RAW 264.7 cell line. Korean J. Intern. Med. 2017, 32, 865–874.
- Kim, C.; Lee, J.M.; Park, S.W.; Kim, K.S.; Lee, M.W.; Paik, S.; Jang, A.S.; Kim, D.J.; Uh, S.; Kim, Y.; et al. Attenuation of Cigarette Smoke-Induced Emphysema in Mice by Apolipoprotein A-1 Overexpression. Am. J. Respir. Cell Mol. Biol. 2016, 54, 91–102.