CYP3A4 is a low specificity isoenzyme of the CYPs family, which contributes to the metabolism of approximately 50% of all marketed drugs. Induction or inhibition of CYP3A4 activity results in the varied oral bioavailability and unwanted drug-drug, drug-food, and drug-herb interactions.
- oral drug delivery
- drug-drug interaction
- bioavailability
- CYP3A4
- BDDCS
- gastrointestinal tract
- P-glycoprotein
- biological barrier
- nutraceutics
- lipid-based nanoparticles
1. Introduction
Failed drug therapy due to unintended drug-drug interactions occurs widely in clinical medicine and could impact drug efficacy and safety [1][2][3]. According to the World Health Organization, the annual financial cost of medically-related harm globally is approximately $42 billion USD [4]. In order to exert a therapeutic effect, drugs must be absorbed via a certain route of administration, such as oral, intravenous, intramuscular, nasal and subcutaneous, among which oral delivery is the most preferred method due to several well-recognized reasons, including convenience, non-invasiveness, extended drug release, suitability for long-acting medication and a long shelf-life [5][6], but the liver and small intestine constitute the main sites of drug metabolism, leading to pharmacokinetic (PK) variability (i.e., different drug concentrations in the blood or at sites of action) [7][8]. Special populations, such as the elderly, children, women, pregnancy, hospitalized patients and even certain ethnicities, are particularly vulnerable to certain prescribed oral medication(s), because their physiological differences (e.g., metabolism) and/or ongoing life circumstances (e.g., polypharmacy, comorbidities) may result in unpredictable drug-drug interactions [9].
Before reaching the target site of action, orally administered drugs encounter a host of obstacles in the gastrointestinal tract (GIT), such as a substantially changing pH in the stomach, upper and lower intestinal segments, extensive enzymatic degradation (e.g., lipase, trypsin, amylase), varied GI motility (e.g., gastric emptying, peristalsis), complex bacterial diversity and physical barriers of the mucus and mucosal layers [10][11]. Among those complex GIT environmental factors, pre-systemic metabolism of Cytochrome P450 (CYPs) enzymes expressed in the intestine and liver is the main factor that significantly contributes to the variability in drug response [12]. Particularly, out of all the CYPs involved in human drug biotransformation, CYP3A4 is the most important oxidation enzyme by virtue of the fact that at least 50% of marketed drugs metabolized by CYPs are metabolized by CYP3A4 [8][13]. The amount and activity of CYP3A4 is considerably regulated by inflammation, fasting state and a broad spectrum of xenobiotics, including top prescribed pharmaceuticals (e.g., midazolam, felodipine), common foods (e.g., grapefruit juice) and widely used herbal medicines (e.g., St. John wort (SJW)) [14][15][16] ( Figure 1 ). However, drug development selects the most effective and safe dose in the study population (not the individual) without any regard for such drug response variability due to CYP3A4 metabolism, as it is impractical to investigate many different doses in different patients. Yet, from a clinical medicine point of view, “one-dose-fits-all” regimen can be potentially dangerous for patients as huge inter- and intra-individual variability in CYP3A4 may lead to varied systemic drug concentrations, in turn, causing unpredictable therapeutic outcomes and intolerable adverse effects [17].
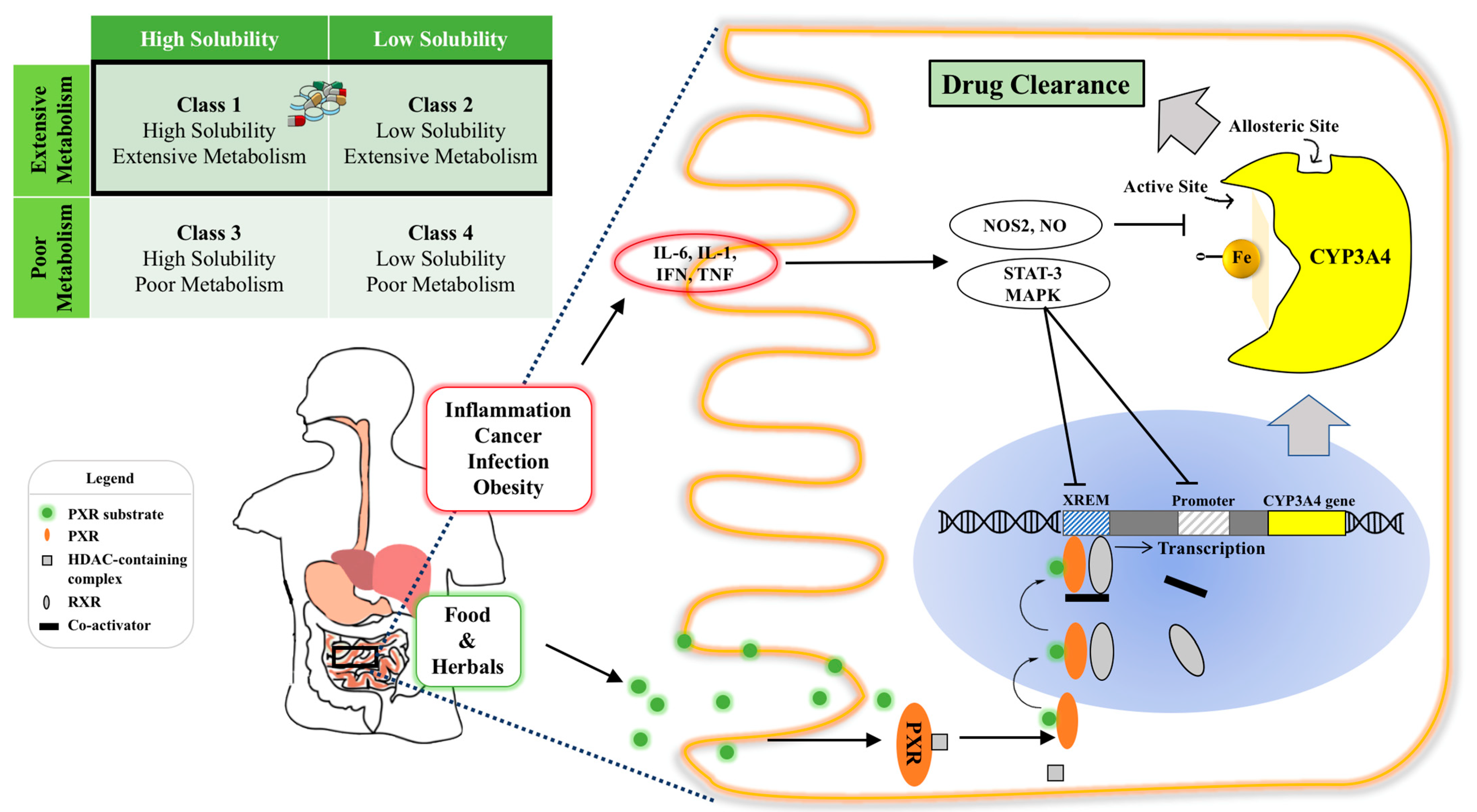
Figure 1. Illustration of intestinal CYP3A4 regulation and the effect of its content and activity variation on orally administered drugs. Top left: CYP3A4 ligands belong to BDDCS Class 1 and Class 2 with varied solubilities and extensive metabolism, highlighted by the black rectangle; Bottom panel: intra-enterocytic CYP3A4 regulation by endogenous factors under disease conditions and xenobiotics from oral intake (e.g., drugs or food constituents). Normally, a PXR ligand enters the enterocyte and binds to PXR intracellularly. This then dimerizes with retinoid X receptor (RXR) and binds to the xenobiotic response enhancer module (XREM) to upregulate the CYP3A4 gene. Systemic inflammatory conditions such as cancer, infection and obesity increase circulating cytokines, such as IL-6, which activate the STAT3-MAPK pathway to downregulate CYP3A4 gene regulation. Intra- and inter-individual CYP3A4 variation can cause varied drug clearance, resulting in undesirable toxicity and ineffective therapy of drugs with a narrow therapeutic index. Abbreviation: RXR, retinoid X receptor; XREM, xenobiotic response enhancer module (XREM); STAT: signal transducer and activator of transcription-3; MAPK: a family of signaling cascade including Jun N-terminal kinase and mitogen-activated protein kinase 1 pathways.
Despite incongruity between industrial and clinical sectors in how to determine drug dosing, for most drugs, the prediction of their in vivo efficacy and toxicity relies on free (i.e., bioavailable) drug concentration at the site of action [18]. However, before reaching systemic circulation, the passage of orally administered drugs through the GIT could be attenuated by the presence of drug transporters (e.g., P-glycoprotein (P-gp)) and metabolizing enzymes (e.g., CYP3A4) in the small intestine. The additional barrier of first-pass metabolism, as the portal vein flows through the liver, also contributes to drug elimination.
2. LNS Strategies of Overcoming Pre-Systemic CYP3A4 Metabolism
| LNS | a | Delivery Mechanism | Nanoformulations | b | Drug Payload | BDDCS Class | c | Study Models | Main Effects | b | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipid NPs | Mucoadhesive | SLN | Cyclosporin A | Class 2 | Young pig |
| [45] | ||||
| Mucoadhesive | VP16-NLC | Etoposide | Class 3 | Rat intestinal membrane, Healthy rat |
| [46] | |||||
| Clathrin-mediated endocytosis | DRD-SLN | Dronedarone hydrochloride | Class 2 | Healthy rat |
| [47] | |||||
| Lymphatic transport via chylomicrons | EFV-SLN | Efavirenz | Class 2 | Chylomicron blocking rat model, Mesenteric lymph duct cannulated rat model |
| [48] | |||||
| Lymphatic transport via chylomicrons | AT-NLC | Atorvastatin | Class 2 | Both High-fat diet treated and health rats |
| [49] | |||||
| Portal vein and lymphatic pathway transport | Darunavir-SLN | Darunavir | Class 2 | Everted rat intestine, Chylomicron blocking rat model, Healthy rats |
| [50] | |||||
| Portal vein and lymphatic pathway transport | AM-SLNs | Asenapine maleate | Class 1 | Caco-2 monolayer, Chylomicron blocking rat model |
| [51] | |||||
| Lymphatic absorption | CLA-SLN | Clarithromycin | Class 3 | Healthy rat |
| [52] | |||||
| Lymphatic absorption | GEN-loaded SLN | Genistein | CYP3A4 inhibitor | In vitro characterization of chylomicrons Caco-2 cells, Ex vivo porcine duodenum |
| [53] | |||||
| Lymphatic transport via chylomicrons | micelles | 5-demethylnobiletin | N/A | Caco-2 monolayer |
| [54] | |||||
| Portal vein and lymphatic pathway transport | CCN | Candesartan cilexetil | Class 4 | Caco-2 monolayer, in situ single-pass intestine perfusion, ligated intestinal loop model, Healthy rats |
| [55] | |||||
| PLN | Mucus penetration | pSLN | Doxorubicin | Class 1 | Caco-2/HT29 co-culture, Everted rat intestine, Intestine loops model, Healthy rat |
| [56] | ||||
| Mucoadhesive | Chitosan coated liposome | Alendronate | Class 3 | Caco-2 monolayer, Healthy rat |
| [57] | |||||
| Enterocyte adhesive via WGA-lectin binding | LPSN | Paclitaxel | Class 2 | A549 cells, Healthy rats |
| [58] | |||||
| M-cell phagocytosis, TJ opening, and caveola-mediated endocytosis | HACC-DTX-SLN | Docetaxel | Class 2 | Caco-2 monolayer, FAE monolayer, Healthy rat |
| [59] | |||||
| Lymphatic uptake | NCC-SLN | Curcumin | CYP3A4 inhibitor | Healthy rat |
| [60] | |||||
| pH responsive drug release (i.e., pH 1.2 and pH 7.4) | EuC-NLS | Alendronate sodium | Class 3 | Healthy rabbit |
| [61] | |||||
| pH-responsive drug release, (i.e., pH>7.0) | IRSLNF3 | Irinotecan hydrochloride trihydrate | Class 1 | Healthy mice, HT-29 bearing mice |
| [62] | |||||
| Targeting MCT1 transport | DTX-ACSL-Lip | Docetaxel | Class 2 | 4T1 and Caco-2 cells, Healthy rats |
| [63] | |||||
| Targeting ASBT in the distal ileum | DSLN-CSG | Docetaxel | Class 2 | Lymph fistula rat model, Healthy rats and tumor bearing mice |
| [64] | |||||
| Targeting VB | 12 | mediated endocytosis | H/VC-LPN | Curcumin | CYP3A4 inhibitor | Caco-2/HT29-MTX co-culture, Healthy mice & rats |
| [65] |
3. Conclusions
References
- Coleman, J.J. Prescribing in 2019: What are the safety concerns? Expert Opin. Drug Saf. 2019, 18, 69–74.
- Pang, K.S.; Rodrigues, A.D.; Peter, R.M. Enzyme- and Transporter-Based Drug–Drug Interactions: Progress and Future Challenges, 1st ed.; Springer Science Business Media LLC: New York, NY, USA, 2010.
- Polasek, T.M.; Shakib, S.; Rostami-Hodjegan, A. Precision dosing in clinical medicine: Present and future. Expert Rev. Clin. Pharmacol. 2018, 11, 743–746.
- WHO. Global Patient Safety Challenge: Medication Without Harm; World Health Organisation: Geneva, Switzerland, 2017; Available online: https://www.who.int/initiatives/medication-without-harm (accessed on 11 May 2021).
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129.
- Traverso, G.; Langer, R. Perspective: Special delivery for the gut. Nature 2015, 519, S19.
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.G.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Ceulemans, J.; Corsetti, M.; Griffin, B.T.; Grimm, M.; et al. Impact of gastroin-testinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812–105845.
- Uetrecht, J.P.; Trager, W. Drug Metabolism: Chemical and Enzymatic Aspects; CRC Press: Boca Raton, FL, USA, 2007.
- Grimsrud, K.N.; Sherwin, C.M.T.; Constance, J.E.; Tak, C.; Zuppa, A.F.; Spigarelli, M.G.; Mihalopoulos, N.L. Special population considerations and regulatory affairs for clinical research. Clin. Res. Regul. Aff. 2015, 32, 45–54.
- Gamboa, J.M.; Leong, K.W. In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 800–810.
- Galipeau, H.; Verdu, E.F. The complex task of measuring intestinal permeability in basic and clinical science. Neurogastroenterol. Motil. 2016, 28, 957–965.
- Nebert, D.W.; Russell, D. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162.
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141.
- Morgan, E.T.; Goralski, K.B.; Piquette-Miller, M.; Renton, K.W.; Robertson, G.R.; Chaluvadi, M.R.; Charles, K.A.; Clarke, S.J.; Kacevska, M.; Liddle, C.; et al. Regulation of drug-metabolizing enzymes and trans-porters in infection, inflammation, and cancer. Drug Metab. Dispos. 2008, 36, 205–216.
- Harris, R.Z.; Jang, G.R.; Tsunoda, S. Dietary Effects on Drug Metabolism and Transport. Clin. Pharmacokinet. 2003, 42, 1071–1088.
- Lammers, L.A.; Achterbergh, R.; Romijn, J.A.; Mathôt, R.A.A. Nutritional Status Differentially Alters Cytochrome P450 3A4 (CYP3A4) and Uridine 5′-Diphospho-Glucuronosyltransferase (UGT) Mediated Drug Metabolism: Effect of Short-Term Fasting and High Fat Diet on Midazolam Metabolism. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 751–767.
- Dresser, G.K.; Spence, J.D.; Bailey, D.G. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cyto-chrome p450 3a4 inhibition. Clin. Pharmacokinet. 2000, 38, 41–57.
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939.
- Cheng, L.; Wong, H. Food effects on oral drug absorption: Application of physiologically-based pharmacokinetic modeling as a predictive tool. Pharmaceutics 2020, 12, 672.
- Custodio, J.M.; Wu, C.-Y.; Benet, L.Z. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv. Drug Deliv. Rev. 2008, 60, 717–733.
- Singh, B.N. Effects of Food on Clinical Pharmacokinetics. Clin. Pharmacokinet. 1999, 37, 213–255.
- O’Shea, J.P.; Holm, R.; O’Driscoll, C.M.; Griffin, B.T. Food for thought: Formulating away the food effect—A PEARRL review. J. Pharm. Pharmacol. 2018, 71, 510–535.
- Huizinga, J.D.; Chen, J.-H.; Zhu, Y.F.; Pawelka, A.J.; McGinn, R.; Bardakjian, B.L.; Parsons, S.; Kunze, W.A.; Wu, R.Y.; Bercik, P.; et al. The origin of segmentation motor activity in the intestine. Nat. Commun. 2014, 5, 1–11.
- Food-Effect Bioavailability and Fed Bioequivalence Studies. Available online: http://www.fda.gov/cder/guidance/index.htm (accessed on 3 May 2021).
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496.
- Zhang, D.; Hop, C.E.; Patilea-Vrana, G.; Gampa, G.; Seneviratne, H.; Unadkat, J.D.; Kenny, J.R.; Nagapudi, K.; Di, L.; Zhou, L.; et al. Drug Concentration Asymmetry in Tissues and Plasma for Small Molecule–Related Therapeutic Modalities. Drug Metab. Dispos. 2019, 47, 1122–1135.
- Drug Bioavailability. Available online: https://www.merckmanuals.com/professional/clinical-pharmacology/pharmacokinetics/drug-bioavailability (accessed on 7 June 2021).
- Mueller, E.A.; Jm, K.; Kutz, K. Minor influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of cy-closporine. Transpl. Proc. 1994, 26, 2957–2958.
- Mueller, E.A.; Kovarik, J.M.; van Bree, J.B.; Grevel, J.; Lucker, P.W.; Kutz, K. Influence of a fat-rich meal on the pharmacokinetics of a new oral for-mulation of cyclosporine in a crossover comparison with the market formulation. Pharm. Res. 1994, 11, 151–155.
- Nanjwade, B.K.; Patel, D.J.; Udhani, R.A.; Manvi, F.V. Functions of Lipids for Enhancement of Oral Bioavailability of Poorly Water-Soluble Drugs. Sci. Pharm. 2011, 79, 705–727.
- Humberstone, A.J.; Charman, W.N. Lipid-based vehicles for the oral delivery of poorly water-soluble drugs. Adv. Drug Deliv. Rev. 1997, 25, 103–128.
- Poovi, G.; Damodharan, N. Lipid nanoparticles: A challenging approach for oral delivery of BCS Class-II drugs. Futur. J. Pharm. Sci. 2018, 4, 191–205.
- Ganesan, P.; Narayanasamy, D. Lipid nanoparticles: Different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain. Chem. Pharm. 2017, 6, 37–56.
- He, H.; Lu, Y.; Qi, J.; Zhu, Q.; Chen, Z.; Wu, W. Adapting liposomes for oral drug delivery. Acta Pharm. Sin. B 2018, 9, 36–48.
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Self-nano-emulsifying drug delivery systems: An update of the biopharmaceutical aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133.
- Zhang, R.X.; Ahmed, T.; Li, L.Y.; Li, J.; Abbasi, A.Z.; Wu, X.Y. Design of nanocarriers for nanoscale drug delivery to enhance cancer treatment using hybrid polymer and lipid building blocks. Nanoscale 2016, 9, 1334–1355.
- Zhang, R.X.; Wong, H.L.; Xue, H.Y.; Eoh, J.Y.; Wu, X.Y. Nanomedicine of synergistic drug combinations for cancer therapy – Strategies and perspectives. J. Control. Release 2016, 240, 489–503.
- Rezhdo, O.; Speciner, L.; Carrier, R. Lipid-associated oral delivery: Mechanisms and analysis of oral absorption enhancement. J. Control. Release 2016, 240, 544–560.
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248.
- Patel, R.; Barker, J.; ElShaer, A. Pharmaceutical Excipients and Drug Metabolism: A Mini-Review. Int. J. Mol. Sci. 2020, 21, 8224.
- Ren, X.; Mao, X.; Cao, L.; Xue, K.; Si, L.; Qiu, J.; Schimmer, A.D.; Li, G. Nonionic surfactants are strong inhibitors of cytochrome P450 3A biotransformation activity in vitro and in vivo. Eur. J. Pharm. Sci. 2009, 36, 401–411.
- Ren, X.; Mao, X.; Si, L.; Cao, L.; Xiong, H.; Qiu, J.; Schimmer, A.; Li, G. Pharmaceutical excipients inhibit cytochrome P450 activity in cell free systems and after systemic administration. Eur. J. Pharm. Biopharm. 2008, 70, 279–288.
- Xu, Y.; Shrestha, N.; Préat, V.; Beloqui, A. Overcoming the intestinal barrier: A look into targeting approaches for improved oral drug delivery systems. J. Control. Release 2020, 322, 486–508.
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124.
- Müller, R.; Runge, S.; Ravelli, V.; Mehnert, W.; Thünemann, A.; Souto, E. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int. J. Pharm. 2006, 317, 82–89.
- Zhang, T.; Chen, J.; Zhang, Y.; Shen, Q.; Pan, W. Characterization and evaluation of nanostructured lipid carrier as a vehicle for oral delivery of etoposide. Eur. J. Pharm. Sci. 2011, 43, 174–179.
- Gambhire, V.M.; Gambhire, M.S.; Ranpise, N.S. Solid Lipid Nanoparticles of Dronedarone Hydrochloride for Oral Delivery: Optimization, In Vivo Pharmacokinetics and Uptake Studies. Pharm. Nanotechnol. 2019, 7, 375–388.
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446.
- Elmowafy, M.; Ibrahim, H.M.; Ahmed, M.A.; Shalaby, K.; Salama, A.; Hefesha, H. Atorvastatin-loaded nanostructured lipid carriers (NLCs): Strategy to overcome oral delivery drawbacks. Drug Deliv. 2017, 24, 932–941.
- Bhalekar, M.R.; Upadhaya, P.; Madgulkar, A.R.; Kshirsagar, S.J.; Dube, A.; Bartakke, U.S. In-vivo bioavailability and lymphatic uptake evaluation of lipid nanoparticulates of darunavir. Drug Deliv. 2015, 23, 2581–2586.
- Patel, M.; Mundada, V.; Sawant, K. Enhanced intestinal absorption of asenapine maleate by fabricating solid lipid nanoparticles using TPGS: Elucidation of transport mechanism, permeability across Caco-2 cell line and in vivo pharmacokinetic studies. Artif. Cells Nanomedicine Biotechnol. 2019, 47, 144–153.
- Sharma, M.; Gupta, N.; Gupta, S. Implications of designing clarithromycin loaded solid lipid nanoparticles on their pharma-cokinetics, antibacterial activity and safety. RSC Advances 2016, 6, 76621–76631.
- Obinu, A.; Burrai, G.; Cavalli, R.; Galleri, G.; Migheli, R.; Antuofermo, E.; Rassu, G.; Gavini, E.; Giunchedi, P. Transmucosal Solid Lipid Nanoparticles to Improve Genistein Absorption via Intestinal Lymphatic Transport. Pharmaceutics 2021, 13, 267.
- Yao, M.; McClements, D.J.; Zhao, F.; Craig, R.W.; Xiao, H. Controlling the gastrointestinal fate of nutraceutical and pharmaceutical-enriched lipid nanoparticles: From mixed micelles to chylomicrons. NanoImpact 2017, 5, 13–21.
- Gao, F.; Zhang, Z.; Bu, H.; Huang, Y.; Gao, Z.; Shen, J.; Zhao, C.; Li, Y. Nanoemulsion improves the oral absorption of can-desartan cilexetil in rats: Performance and mechanism. J. Control. Release 2011, 149, 168–174.
- Yuan, H.; Chen, C.-Y.; Chai, G.; Du, Y.-Z.; Hu, F.-Q. Improved Transport and Absorption through Gastrointestinal Tract by PEGylated Solid Lipid Nanoparticles. Mol. Pharm. 2013, 10, 1865–1873.
- Han, H.-K.; Shin, H.-J.; Ha, D.H. Improved oral bioavailability of alendronate via the mucoadhesive liposomal delivery system. Eur. J. Pharm. Sci. 2012, 46, 500–507.
- Pooja, D.; Kulhari, H.; Kuncha, M.; Rachamalla, S.S.; Adams, D.; Bansal, V.; Sistla, R. Improving Efficacy, Oral Bioavailability, and Delivery of Paclitaxel Using Protein-Grafted Solid Lipid Nanoparticles. Mol. Pharm. 2016, 13, 3903–3912.
- Shi, L.-L.; Xie, H.; Lu, J.; Cao, Y.; Liu, J.-Y.; Zhang, X.-X.; Zhang, H.; Cui, J.-H.; Cao, Q.-R. Positively Charged Surface-Modified Solid Lipid Nanoparticles Promote the Intestinal Transport of Docetaxel through Multifunctional Mechanisms in Rats. Mol. Pharm. 2016, 13, 2667–2676.
- Baek, J.S.; Cho, C.W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioa-vailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140.
- Hosny, K.M.; Ahmed, O.A.A.; Al-Abdali, R.T. Enteric-coated alendronate sodium nanoliposomes: A novel formula to overcome barriers for the treatment of osteoporosis. Expert Opin. Drug Deliv. 2013, 10, 741–746.
- Rajpoot, K.; Jain, S.K. Oral delivery of ph-responsive alginate microbeads incorporating folic acid-grafted solid lipid nano-particles exhibits enhanced targeting effect against colorectal cancer: A dual-targeted approach. Int. J. Biol. Macromol. 2020, 151, 830–844.
- Guo, X.; Zhang, J.; Cai, Q.; Fan, S.; Xu, Q.; Zang, J.; Yang, H.; Yu, W.; Li, Z.; Zhang, Z. Acetic acid transporter-mediated, oral, multifunctional polymer liposomes for oral delivery of docetaxel. Colloids Surf. B Biointerfaces 2020, 198, 111499.
- Kim, K.S.; Youn, Y.S.; Bae, Y.H. Immune-triggered cancer treatment by intestinal lymphatic delivery of docetaxel-loaded nanoparticle. J. Control. Release 2019, 311-312, 85–95.
- Liu, Y.; Jiang, Z.F.; Hou, X.F.; Xie, X.M.; Shi, J.P.; Shen, J.Y.; He, Y.Z.; Wang, Z.; Feng, N.P. Functional lipid polymeric nanoparticles for oral drug delivery: Rapid mucus penetration and improved cell entry and cellular transport. Nanomed. Nanotechnol. Biol. Med. 2019, 21, 102075.
- Benet, L.Z.; Broccatelli, F.; Oprea, T. BDDCS Applied to Over 900 Drugs. AAPS J. 2011, 13, 519–547.