Signal transducer and activator of transcription 3 (STAT3) is a critical transcription factor that has been firmly associated with colorectal cancer (CRC) initiation and development. STAT3 mediates key inflammatory mechanisms in colitis-associated cancer, becomes excessively activated in CRC, and enhances cancer cell proliferation, tumor growth, angiogenesis, invasion, and migration. STAT3 hyperactivation in malignant cells, surrounding immune cells and cancer-associated fibroblasts, mediates inhibition of the innate and adaptive immunity of the tumor microenvironment, and, therefore, tumor evasion from the immune system.
- STAT3
- colorectal cancer
- colorectal cancer inflammation
- colorectal cancer immunotherapy
- colorectal cancer resistance
1. Introduction
2. STAT3 Structure and Regulation of Activity in Normal Cells and Disease
The STAT family consists of the following seven structurally and functionally associated proteins: STAT1, STAT2, STAT3, STAT4, STAT5a and STAT5b, and STAT6. STATs function as cytoplasmic signal transducers and regulators of gene transcription in the nucleus. These molecular events occur following cytokine and growth factor stimuli, and they are critical for the regulation of innate and acquired host immune responses [7][8][12,13]. STAT3 was firstly identified as a component of IL-6 signaling, which co-immunoprecipitates as a phosphoprotein in a complex with acute phase response factor (APRF) in HepG2 hepatoma cells [9][14]. It is an 88-kDa protein that harbors a conserved Src homology 2 (SH2) domain within its primary amino acid sequence [10][15]. Cytokines bind to classic cytokine receptors (e.g., IL-10), and membrane-bound and soluble receptors (e.g. IL-6, IL-11, and IL-23). Growth factors bind to their respective growth factor receptors, and peptide hormones bind to their respective G-protein-coupled receptors (GPCR)s (Figure 1) [10][11][15,16]. Upon ligand binding, the receptors mainly activate the Janus kinase (JAK) family of kinases, which trans-phosphorylate each other. JAKs further phosphorylate the C-terminus of the receptor subunit. The latter event attracts STAT3, with its SH2 domain, to a related docking site in the cytoplasmic tail of the activated receptor subunit. This leads to STAT3 phosphorylation on the Y705 tyrosine residue.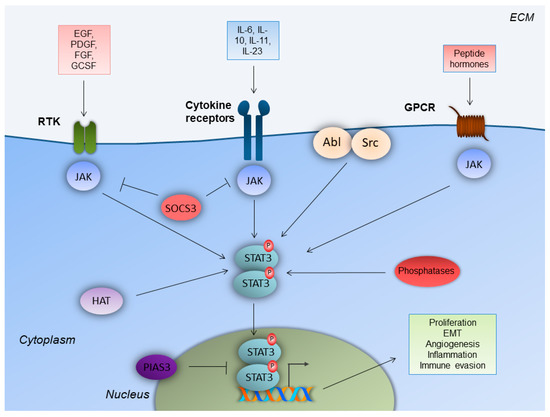
3. Mechanisms of STAT3 Implication in Tumorigenesis
The canonical STAT3 pathway that functions transiently and is strictly regulated in normal cells, has been documented to be persistently activated in a variety of solid and hematological malignancies [17][32]. STAT3 is also activated by non-canonical pathways in tumorigenesis, which involve STAT3 phosphorylation in a (Ser)727 alternative site of the C-terminus, by mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), or protein kinase C (PKC) pathways. Constitutively activated STAT3 translocates to the nucleus and enhances gene transcription, which favors cell proliferation, resistance to apoptosis, survival, angiogenesis, tumor-promoting inflammation, tumor-mediated immune evasion, and, ultimately, invasion and metastasis. Recent evidence demonstrates that STAT3 promotes tumorigenesis through additional processes, including inflammation-promoting carcinogenesis, obesity and/or metabolism, CSCs, and pre-metastatic niche formation [18][33].
Aberrant STAT3 activation is mainly attributed to the excessive production of cytokines and growth factors in the tumor microenvironment (TM), high expression of protein tyrosine kinases, and epigenetic suppression of negative regulators of STAT3, such as SOCS3 and protein tyrosine phosphatases [19][34]. Particularly, the activation of the IL-6/JAK/STAT3 core axis is observed in many types of cancer. IL-6 is overexpressed in the TM and stimulates the JAK/STAT3 pathway, which is a molecular event that is associated with poor prognosis for cancer patients. JAK2 is specifically activated in myeloproliferative disorders and activates STAT3 [20][35]. IL-6 plays a protective role for cancer cells, against DNA damage, apoptosis, and oxidative stress [21][36]. Regarding angiogenesis, STAT3 is activated in cancer cells by reactive oxygen species (ROS), mechanistic target of rapamycin complex 1 (mTORC1) and/or IL-6, and further induces hypoxia-inducible factor 1-alpha (HIF-1a) activation during hypoxia. HIF-1a induces the transcription of vascular endothelial growth factor (VEGF), which, in turn, affects the surrounding endothelial cells. This event activates, once again, STAT3 via the VEGF receptor (VEGFR). In endothelial cells, STAT3, along with HIF-1a and sp1, promotes the transcription of genes that mediate the cell growth, survival, and migration of endothelial cells, therefore promoting angiogenesis [22][37].
4. Mechanisms of STAT3 Engagement in Colorectal Tumorigenesis
4.1. STAT3 Expression and Regulation in Colorectal Cancer Cells
The immunohistochemical evaluation of p-STAT3 in 108 CRC cases demonstrated cytoplasmic and nuclear expression, positive correlation of pSTAT3 expression, with the depth of the tumor invasion, Dukes, TNM stage, and reduced overall survival [23][40]. STAT3-enhanced activation in CRC occurs from IL-6 in the serum and the tumor microenvironment, from growth factors and growth factor receptors, tyrosine kinases (Src, Bcr-Abl), and loss-of-function mutations for the STAT3 suppressors/inhibitors (phosphatases, SOCS, PIAS) [20][35].
Beyond direct STAT3 regulation by upstream molecules, STAT3 is also activated by the suppression of STAT3 negative regulators, such as PTPRT [24][41]. PTPRT functions as a tumor suppressor gene, and is mutated in colon, lung, stomach, and skin (melanoma) cancers. In CRC, it is the most frequently mutated tyrosine phosphatase among the tyrosine phosphatome that was examined in 18 CRCs. PTPRT presents five mutations that decrease its activity [25][26][42,43]. STAT3 has been identified as a substrate of PTPRT. The epigenetic suppression (promoter hypermethylation) of PTPRT occurs in several human cancers, but most notably in CRC (78.7%, 289/367 tumors analyzed) [27][44].
4.2. STAT3 in Invasion and Metastasis
STAT3 is implicated in the invasion and metastasis of CRC cells, through various mechanisms. In CRC cells, the IL-6/STAT3 axis promotes EMT and, therefore, the CRC aggressive phenotype via the STAT3-dependent transcriptional upregulation of Fos-related antigen-1 (Fra-1) TF. Fra-1 belongs to the Fos family of TFs, and is associated with phenotype invasive transformation and EMT in various types of cancer. In a 229 CRC patient cohort, Fra-1 expression was positively correlated with an increased depth of invasion, lymph node, and liver metastases. Fra-1 was also expressed according to IL-6 and pSTAT3 expression. In CRC cells, STAT3 needs to be both phosphorylated and acetylated to bind to the Fra-1 promoter and induce Fra-1 transcription [28][48].
MiR-34 is known to suppress EMT. The MiR-34 gene is a target of activated STAT3, which causes transcriptional repression of miR-34, and therefore induces EMT promotion in CRC cells and tumors [29][49]. Furthermore, the p53-inducible miR-34a control of the tyrosine kinase colony-stimulating factor 1 receptor is alleviated in CRC, through a feedback loop that is mediated by STAT3 [30][50]. STAT3 is also involved by enhancing the acquisition of EMT and cancer stem cells traits by CRC cells, through transcriptional induction of NANOG TF.
4.3. STAT3 in Angiogenesis
During hypoxia, STAT3 is activated in cancer cells by ROS, mTORC1, and/or IL-6, and further induces HIF-1a. This transcription factor induces the transcription of VEGF. This event affects the surrounding endothelial cells, in order to activate, once again, STAT3 via VEGFR. In endothelial cells, STAT3, along with HIF-1a and sp1, promotes the transcription of genes that mediate cell growth, survival, and migration of ECS, therefore angiogenesis [22][37]. Corticotrophin-releasing hormone (CRH) and its receptor CRHR contribute to CRC cells proliferation, and promote angiogenesis.
4.4. STAT3 in Tumor-Promoting Inflammation
5. STAT3 in Colorectal Cancer Treatment
We already know that STAT3 inhibition in vitro suppresses tumor growth and promotes apoptosis in cancer cells. STAT3 integrates the oncogenic signaling of multiple upstream kinase molecules. Therefore, STAT3 inhibition is a promising therapeutic strategy that could abrogate the oncogenic impact of several upstream activated kinase molecules. Although JAK1/2 inhibition has favorable effects in JAK/STAT-driven hematological malignancies, agents that target STAT3 are still in the early phases of development. Strategies targeting STAT3 are categorized into the following groups: (1) inhibitors of the SH2 domain or STAT3 dimerization, (2) upstream tyrosine kinase inhibitors, (3) STAT3 pathway oligonucleotides, (4) inhibitors of STAT3–DNA binding, and (5) mimics of STAT3 negative regulators [17][32]. There is a number of STAT3 inhibitors with promising efficacy in preclinical studies, which have entered clinical trials (Table 1) [6][10].| Identifier | Status | Title | Condition | Phase | Drug |
|---|---|---|---|---|---|
| NCT03647839 | Active, not recruiting | Modulation Of The Tumour Microenvironment Using Either Vascular Disrupting Agents or STAT3 Inhibition in Order to Synergise With PD1 Inhibition in Microsatellite Stable, Refractory Colorectal Cancer | Colorectal Cancer Metastatic | II | BBI608 |
| NCT02983578 | Active, not recruiting | Danvatirsen and Durvalumab in Treating Patients With Advanced and Refractory Pancreatic, non-small cell Lung Cancer, and Mismatch Repair Deficient Colorectal Cancer | Advanced Colorectal Carcinoma Mismatch Repair Deficiency Refractory Colorectal Carcinoma Stage III Colorectal Cancer AJCC v8 Stage IIIA Colorectal Cancer AJCC v8 Stage IIIB Colorectal Cancer AJCC v8 Stage IIIC Colorectal Cancer AJCC v8 Stage IV Colorectal Cancer AJCC v8 Stage IVA Colorectal Cancer AJCC v8 Stage IVB Colorectal Cancer AJCC v8 Stage IVC Colorectal Cancer AJCC v8 |
II | Danvatirsen |
| NCT03195699 | Recruiting | Oral STAT3 Inhibitor, TTI-101, in Patients With Advanced Cancers | Colorectal Cancer Advanced Cancer |
I | TTI-101 |
| NCT03522649 | Recruiting | A Phase III Clinical Study of Napabucasin (GB201) Plus FOLFIRI in Adult Patients With Metastatic Colorectal Cancer | Previously Treated Metastatic Colorectal Cancer | III | BBI608 |
| NCT02753127 | Completed | A Study of Napabucasin (BBI-608) in Combination With FOLFIRI in Adult Patients With Previously Treated Metastatic Colorectal Cancer (CanStem303C) | Colorectal cancer | III | BBI608 |
| NCT01776307 | Completed | A Study of BBI608 in Adult Patients With Advanced Colorectal Cancer | Colorectal Cancer | II | BBI608 |
| NCT03522649 | Recruiting | A Phase III Clinical Study of Napabucasin (GB201) Plus FOLFIRI in Adult Patients With Metastatic Colorectal Cancer | Previously Treated Metastatic Colorectal Cancer | III | BBI608 |
| NCT02641873 | Completed | A Study of BBI608 Administrated With FOLFIRI + Bevacizumab in Adult Patients With Metastatic Colorectal Cancer | Metastatic Colorectal Cancer | I | BBI608 |
| NCT03647839 | Active, not recruiting | Modulation Of The Tumour Microenvironment Using Either Vascular Disrupting Agents or STAT3 Inhibition in Order to Synergise With PD1 Inhibition in Microsatellite Stable, Refractory Colorectal Cancer (MODULATE) | Colorectal Cancer Metastatic | II | BBI608 |
| NCT01830621 | Completed | BBI608 and Best Supportive Care vs. Placebo and Best Supportive Care in Pretreated Advanced Colorectal Carcinoma | Colorectal Carcinoma | III | BBI608 |