Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Pasquale Esposito and Version 2 by Camila Xu.
Myostatin and activin-A are two of thirty-three members of the TGF-β family. Here, we briefly describe their basic biology and main functions.
- myostatin
- activin A
- activin receptors
- atherosclerosis
1. Myostatin/Activin-A Signaling: Basic Biology and Functions
1. Myostatin Biology
Myostatin (Mstn)/GDF8 is synthesized as an inactive 375 KDa precursor (pre-pro Mstn), bearing at N-terminal domain a hydrophobic core of 24 amino acids (signal-peptide) and at the C-terminal region, 9 cysteine residues and a proteolytic processing site RSSR (Arg-Ser-Arg-Arg) recognized by furin, a calcium-dependent serine protease [1]. The proteolytic cleavage produces a 36/40 kDa latency-associated peptide (LAP) and a 12.5/26 kDa mature peptide, corresponding to a C-terminal monomer or dimer, respectively, that is the biologically active molecule. The non-covalent interaction with LAP retains Mstn in a latent state that cannot engage its receptor [2]. Only the subsequent proteolytic cleavage by BMP1/Tolloid family metalloproteases releases active Mstn [3].
1.1. Mstn Signaling and Functions
Mstn signals through a heteromeric complex of type I and type II receptors that are transmembrane proteins of approximately 55 and 70 KDa, respectively [4] (Figure 1), differing in level of sequence homology within the kinase domains and the presence of a highly conserved glycine–serine-rich (GS) domain in the cytoplasmic region of the type I receptor [5]. Mstn receptor engagement and its downward signaling are negatively regulated by the Mstn prodomain and by antagonists, including follistatin (FSTL) [6], follistatin-like 3 (FSTL3), GDF-associated serum protein-1 (GASP-1) [7] and Cripto, a small glycosylphosphatidylinositol-linked membrane-associated protein (GPI)-anchored signaling protein [8][9]. Firstly, Mstn binds to activin receptor IIB (ActRIIB) and then, this complex phosphorylates a type I activin receptor-like kinase-4 (ALK4) (myogenic cells), or -5 (ALK5) (nonmyogenic cells) [10]. This, in turn, phosphorylates small mothers against decapentaplegic (SMAD)2 and SMAD3 which recruit SMAD4, forming a SMAD2/3/4 complex that enters into the nucleus, regulating gene expression both positively and negatively [11]. SMAD7 works as a negative feedback inhibitor for the SMAD signal pathway [12]. Mainly, the canonical SMAD pathway mediates the effects of Mstn on myogenesis and muscle atrophy, upregulating the ubiquitin-proteasome system (UPS) synergistically with forkhead box transcription factors (FOXOs) and inhibiting the anabolic PI3-K/AKT/mTOR pathway [13]. However, Mstn can activate mitogen-activated protein kinases (MAPKs) that inhibit myoblast proliferation and differentiation [14] and promote an inflammatory milieu stimulating the expression of inflammatory cytokines [15]. On the other hand, inflammatory cytokines such as TNFα can induce Mstn expression through NF-kB [16]. Differently, Mstn has inhibitory effects on the Wnt/β-catenin pathway, thereby blunting satellite cell proliferation [17]. Mstn can exert pleiotropic regulatory effects. It has been found primarily expressed by animal and human skeletal muscle cells, where it limits muscle growth [18], inhibiting myogenesis and contributing to muscle loss through the activation of proteolysis and autophagy, mainly by upregulation of the UPS [19][20]. The negative effects of Mstn on muscle growth have been involved in the pathogenesis of age-related sarcopenia [21], as well as in cachexia associated with cancer [22], chronic kidney disease [23], and heart failure [24]. However, beyond regulating skeletal muscle growth, subsequent evidence has proved that Mstn is implicated in many physiological and pathological processes at the systemic level, including energy metabolism, the development of obesity, and insulin resistance [25][26]. Moreover, Mstn may play a role in the pathogenesis of diabetic nephropathy and heart failure [27]. In human proximal tubule cells, Mstn promotes proximal tubule activation and intracellular reactive oxygen species (ROS) release by upregulating NADPH oxidase. In patients with diabetic nephropathy, Mstn is associated with tubulointerstitial infiltrates and fibrotic areas [28]. The effects of Mstn on bone are less studied. Studies of Mstn-knockout mice revealed that early bone regeneration and inhibition of Mstn leads to an increase in osteogenesis. Mstn-/- mice showed an increase in density, strength, and bone mineralization [29]. In clinical studies, inhibition of Mstn increased the osteogenic potential and bone mineralization in patients with diabetes mellitus [30].
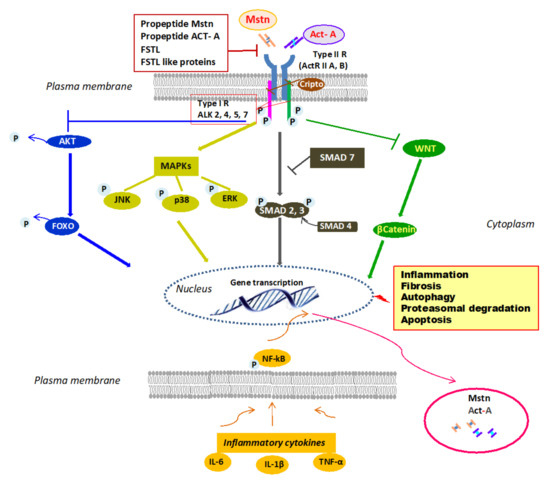
Figure 1. Mstn/Act-A cell signaling. Mstn/Act-A bind to ActRIIB/A on the plasma membrane, which leads to activation by phosphorylation of type-1 ALK 2, 4, 5, or 7. This in turn induces phosphorylation of SMAD2 and SMAD3 and the involvement of SMAD4 into a SMAD complex that translocates into the nucleus and elicits transcription of gene targets. SMAD7 inhibits SMAD pathway. Mstn and Act-A block AKT phosphorylation and consequently, dephosphorylated FOXO can enter the nucleus and promote the transcription and expression of atrophy specific genes. Mstn and Act-A can also signal through MAPK activation and WNT inhibition. The binding to the receptor is controlled by Mstn–Act-A propeptides and by circulating antagonists. Lastly, the ActRII/ActRIB receptor complex is inhibited by Cripto, a small GPI-anchored signaling protein. Inflammatory milieu contributes to Mstn and Act-A expression through NF-kB. Abbreviations: Act-A = activin A, ActRIIB/A = activin receptor type IIB, IIA; ALK = type-1 activin receptor serine kinase2, 4, 5, or 7; Mstn = myostatin; ERK = extracellular signal-regulated kinases; FOXO = forkhead box transcription factor; FSTL = follistatin; GPI = glycosylphosphatidylinositol-linked membrane-associated protein; IL-6 = interleukin-6; IL-1β = interleukin-1β; JNK = c-Jun N-terminal kinase; MAPKs = mitogen-activated protein kinases; NF-KB = nuclear factor kappa-light-chain-enhancer of activated B cells; P = phosphorylated; SMAD= small mothers against decapentaplegic; TNF-α = tumor necrosis factor-α; WNT = portmanteau of Wingless and integrated (WNT)/b-catenin pathway.
2. Activin-A Biology
Activin-A (Act-A) is a homodimer composed of 2 βA subunits linked with disulfide bonds. Act-A is produced as a larger precursor that is cleaved at amino-terminal sequence, releasing the mature 25 KDa carboxy-terminal bioactive ligand [31]. Differently from Mstn, which circulates as a latent precursor complex, the activin propeptide sequence has a weak affinity for the mature dimer, is easily displaced, and does not interfere with receptor binding. The subunits of Act-A are the products of the inhibin beta A gene, because of their original identification as subunits of the gonadal hormone, inhibin [32].
2.1. Act-A Signaling and Functions
Similarly to Mstn, Act-A signals by binding with high affinity to activin receptor type llA (ActRllA) or less so to activin receptor type IIB followed by the recruitment of the ActRI (ALK4, ALK7, or ALK2) [33] (Figure 1). Subsequently, the canonical SMAD pathway is activated. However, Act-A can induce MAPKs, which affect cell migration and differentiation [34][35], and controls the Wnt signaling pathway involved in developmental and injury processes [36]. Act-A action is regulated by several molecules. At the extracellular level, FSTL binds to Act-A with high affinity and prevents the engagement of type II receptors [37] and inhibin, binding to betaglycan, a type III TGF-B receptor, and then to ActRII, and competes with Act-A for the receptor site [38]. At the intracellular level, BAMBI (BMP and activin membrane-bound inhibitor), a transmembrane pseudoreceptor structurally similar to type I receptors, inhibits activin signaling [39] due to lacking the intracellular kinase domain. However, Cripto reduces Act-A efficacy, inhibiting the ability of the activin/ActRII complex to recruit the type I receptor, thereby inhibiting the activin downstream signaling pathway [9]. Act-A was first described as gonadal protein stimulating FSH secretion by the pituitary gland [40]. Then, it is expressed in the embryonal ovary, uterus, and testis, and in glands such as the breast and prostate. Interestingly, Act-A is also present in the human placenta, amnion, and chorion, and its levels are elevated in pregnant women [41]. However, apart from exerting diverse biological functions in the reproductive tract, Act-A and its receptors have been fully characterized in virtually all body systems [42]. Act-A is strongly expressed in different compartments of the central nervous system, where it seems to exert neuroprotective effects [43]. In addition, Act-A is present in both developing and adult heart, kidney, lung, and gastrointestinal tract [44]. In the heart, Act-A (and ActRs) may influence cardiomyocyte differentiation and remodeling after different kinds of injuries [45]. In the kidney, Act-A exerts a profibrotic effect, both during organ development and following acute and chronic damage [46][47]. Moreover, Act-A operates in concert with Mstn in negatively regulating muscle growth and may play a significant role in bone remodeling [48]. In bone, Act-A is secreted by osteoblasts and during bone matrix resorption by osteoclasts [49]. Different animal models showed that Act-A induces osteoblastogenesis, osteoclastogenesis, chondrogenesis, and collagen synthesis [50]. Coherently, inhibition of Act-A signaling obtained by administration of soluble ActRIIA, or the use of a ligand trap, was effective in preventing muscle wasting in different mouse models of experimental CKD and promoted osteogenesis and increased bone mass in healthy mice and primates [51][52]. Finally, Act-A modulates innate and adaptive immune mechanisms and mediates inflammatory responses [53]. Most immune cell types, including macrophages and T and B lymphocytes, can produce and respond to Act-A, and in inflammatory conditions, high levels of interleukin (IL)-1β and tumor necrosis factor-alpha (TNF-α) can promote, through NF-kB, Act-A expression and secretion, boosting the inflammatory process [54]. Furthermore, Act-A is proapoptotic in several cells, such as hepatocytes [55], renal proximal tubular cells [56], B cells [57], chronic myeloid leukaemia cells [58], and cardiac myocytes [59]. Finally, recently it has been proved that activin-A is involved in the pathogenesis of vascular calcification and CKD-related mineral bone disorders.[60]
References
- McPherron, A.C.; Lee, S.-J. The transforming growth factor β superfamily. In Growth Factors and Cytokines in Health and Disease; Leroith, D., Bondy, Eds.; JAI: Stamford, CT, USA, 1996; Volume 1, pp. 357–393. ISBN 1874-5687.
- Kusakabe, M.; Cheong, P.-L.; Nikfar, R.; McLennan, I.S.; Koishi, K. The structure of the TGF-beta latency associated peptide region determines the ability of the proprotein convertase furin to cleave TGF-betas. J. Cell Biochem. 2008, 103, 311–320.
- Walker, R.G.; McCoy, J.C.; Czepnik, M.; Mills, M.J.; Hagg, A.; Walton, K.L.; Cotton, T.R.; Hyvönen, M.; Lee, R.T.; Gregorevic, P.; et al. Molecular characterization of latent GDF8 reveals mechanisms of activation. Proc. Natl. Acad. Sci. USA 2018, 115, E866–E875.
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700.
- Attisano, L.; Wrana, J.L.; Montalvo, E.; Massagué, J. Activation of signalling by the activin receptor complex. Mol. Cell Biol. 1996, 16, 1066–1073.
- Amthor, H.; Nicholas, G.; McKinnell, I.; Kemp, C.F.; Sharma, M.; Kambadur, R.; Patel, K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev. Biol. 2004, 270, 19–30.
- Le, V.Q.; Iacob, R.E.; Tian, Y.; McConaughy, W.; Jackson, J.; Su, Y.; Zhao, B.; Engen, J.R.; Pirruccello-Straub, M.; Springer, T.A. Tolloid cleavage activates latent GDF8 by priming the pro-complex for dissociation. EMBO J. 2018, 37, 384–397.
- Guardiola, O.; Lafuste, P.; Brunelli, S.; Iaconis, S.; Touvier, T.; Mourikis, P.; De Bock, K.; Lonardo, E.; Andolfi, G.; Bouché, A.; et al. Cripto regulates skeletal muscle regeneration and modulates satellite cell determination by antagonizing myostatin. Proc. Natl. Acad. Sci. USA 2012, 109.
- Gray, P.C.; Harrison, C.A.; Vale, W. Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 5193–5198.
- Kemaladewi, D.U.; Gorter, D.J.J.; Aartsma-Rus, A.; Ommen, G.; ten Dijke, P.; Hoen, P.A.C.t.; Hoogaars, W.M. Cell-type specific regulation of myostatin signaling. FASEB J. 2012, 26, 1462–1472.
- Hata, A.; Chen, Y.G. TGF-β signaling from receptors to smads. Cold Spring Harb. Perspect. Biol. 2016, 8.
- Zhu, X.; Topouzis, S.; Liang, L.-F.; Stotish, R.L. Myostatin signaling through Smad2, Smad3 and Smad4 is regulated by the inhibitory Smad7 by a negative feedback mechanism. Cytokine 2004, 26, 262–272.
- Goodman, C.A.; McNally, R.M.; Hoffmann, F.M.; Hornberger, T.A. Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 2013, 27, 1946–1957.
- Philip, B.; Lu, Z.; Gao, Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal. 2005, 17, 365–375.
- Hu, S.-L.; Chang, A.-C.; Huang, C.-C.; Tsai, C.-H.; Lin, C.-C.; Tang, C.-H. Myostatin Promotes Interleukin-1β Expression in Rheumatoid Arthritis Synovial Fibroblasts through Inhibition of miR-21-5p. Front. Immunol. 2017, 8, 1747.
- Zhang, L.; Rajan, V.; Lin, E.; Hu, Z.; Han, H.Q.; Zhou, X.; Song, Y.; Min, H.; Wang, X.; Du, J.; et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 1653–1663.
- Steelman, C.A.; Recknor, J.C.; Nettleton, D.; Reecy, J.M. Transcriptional profiling of myostatin-knockout mice implicates Wnt signaling in postnatal skeletal muscle growth and hypertrophy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 580–582.
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N. Engl. J. Med. 2004, 350, 2682–2688.
- McFarlane, C.; Plummer, E.; Thomas, M.; Hennebry, A.; Ashby, M.; Ling, N.; Smith, H.; Sharma, M.; Kambadur, R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J. Cell Physiol. 2006, 209, 501–514.
- Lee, J.Y.; Hopkinson, N.S.; Kemp, P.R. Myostatin induces autophagy in skeletal muscle in vitro. Biochem. Biophys. Res. Commun. 2011, 415, 632–636.
- Patel, V.K.; Demontis, F. GDF11/myostatin and aging. Aging 2014, 6, 351–352.
- Shyh-Chang, N. Metabolic Changes During Cancer Cachexia Pathogenesis. In Translational Research in Breast Cancer: Biomarker Diagnosis, Targeted Therapies and Approaches to Precision Medicine; Song, E., Hu, H., Eds.; Springer: Singapore, 2017; pp. 233–249. ISBN 978-981-10-6020-5.
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516.
- Hoenig, M.R. Hypothesis: Myostatin is a mediator of cardiac cachexia. Int. J. Cardiol. 2008, 124, 131–133.
- Chen, Y.; Ye, J.; Cao, L.; Zhang, Y.; Xia, W.; Zhu, D. Myostatin regulates glucose metabolism via the AMP-activated protein kinase pathway in skeletal muscle cells. Int. J. Biochem. Cell Biol. 2010, 42, 2072–2081.
- Milan, G.; Dalla Nora, E.; Pilon, C.; Pagano, C.; Granzotto, M.; Manco, M.; Mingrone, G.; Vettor, R. Changes in muscle myostatin expression in obese subjects after weight loss. J. Clin. Endocrinol. Metab. 2004, 89, 2724–2727.
- Heineke, J.; Auger-Messier, M.; Xu, J.; Sargent, M.; York, A.; Welle, S.; Molkentin, J.D. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation 2010, 121, 419–425.
- Verzola, D.; Milanesi, S.; Viazzi, F.; Ansaldo, F.; Saio, M.; Garibaldi, S.; Carta, A.; Costigliolo, F.; Salvidio, G.; Barisione, C.; et al. Enhanced myostatin expression and signalling promote tubulointerstitial inflammation in diabetic nephropathy. Sci. Rep. 2020, 10, 1–13.
- Kellum, E.; Starr, H.; Arounleut, P.; Immel, D.; Fulzele, S.; Wenger, K.; Hamrick, M.W. Myostatin (GDF-8) deficiency increases fracture callus size, Sox-5 expression, and callus bone volume. Bone 2009, 44, 17–23.
- Wallner, C.; Jaurich, H.; Wagner, J.M.; Becerikli, M.; Harati, K.; Dadras, M.; Lehnhardt, M.; Behr, B. Inhibition of GDF8 (Myostatin) accelerates bone regeneration in diabetes mellitus type 2. Sci. Rep. 2017, 7, 1–11.
- Vale, W.; Wiater, E.; Gray, P.; Harrison, C.; Bilezikjian, L.; Choe, S. Activins and inhibins and their signaling. Ann. N. Y. Acad. Sci. 2004, 1038, 142–147.
- Mason, A.J.; Niall, H.D.; Seeburg, P.H. Structure of two human ovarian inhibins. Biochem. Biophys. Res. Commun. 1986, 135, 957–964.
- Morianos, I.; Papadopoulou, G.; Semitekolou, M.; Xanthou, G. Activin-A in the regulation of immunity in health and disease. J. Autoimmun. 2019, 104, 102314.
- Lacolley, P.; Regnault, V.; Avolio, A.P. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovasc. Res. 2018, 114, 513–528.
- Garibotto, G.; Esposito, P.; Picciotto, D.; Verzola, D. Activin/myostatin receptor signaling and vascular calcifications in chronic kidney disease: A “liaison dangereuse”? Kidney Res. Clin. Pract. 2019, 38, 407–410.
- Kawakami, T.; Ren, S.; Duffield, J.S. Wnt signalling in kidney diseases: Dual roles in renal injury and repair. J. Pathol. 2013, 229, 221–231.
- Harrison, C.A.; Gray, P.C.; Vale, W.W.; Robertson, D.M. Antagonists of activin signaling: Mechanisms and potential biological applications. Trends Endocrinol. Metab. 2005, 16, 73–78.
- Gray, P.C.; Bilezikjian, L.M.; Vale, W. Antagonism of activin by inhibin and inhibin receptors: A functional role for betaglycan. Mol. Cell Endocrinol. 2002, 188, 254–260.
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massagué, J.; Niehrs, C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485.
- Ling, N.; Ying, S.-Y.; Ueno, N.; Shimasaki, S.; Esch, F.; Hotta, M.; Guillemin, R. Pituitary FSH is released by a heterodimer of the β-subunits from the two forms of inhibin. Nature 1986, 321, 779–782.
- Schneider-Kolsky, M.E.; Manuelpillai, U.; Waldron, K.; Dole, A.; Wallace, E.M. The distribution of activin and activin receptors in gestational tissues across human pregnancy and during labour. Placenta 2002, 23, 294–302.
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin A in mammalian physiology. Physiol. Rev. 2019, 99, 739–780.
- Tretter, Y.P.; Hertel, M.; Munz, B.; ten Bruggencate, G.; Werner, S.; Alzheimer, C. Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat. Med. 2000, 6, 812–815.
- Feijen, A.; Goumans, M.J.; van den Eijnden-van Raaij, A.J. Expression of activin subunits, activin receptors and follistatin in postimplantation mouse embryos suggests specific developmental functions for different activins. Development 1994, 120, 3621–3637.
- Oshima, Y.; Ouchi, N.; Shimano, M.; Pimentel, D.R.; Papanicolaou, K.N.; Panse, K.D.; Tsuchida, K.; Lara-Pezzi, E.; Lee, S.-J.; Walsh, K. Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury. Circulation 2009, 120, 1606–1615.
- Maeshima, A.; Zhang, Y.Q.; Nojima, Y.; Naruse, T.; Kojima, I. Involvement of the activin-follistatin system in tubular regeneration after renal ischemia in rats. J. Am. Soc. Nephrol. 2001, 12, 1685–1695.
- Yamashita, S.; Maeshima, A.; Kojima, I.; Nojima, Y. Activin A Is a Potent Activator of Renal Interstitial Fibroblasts. J. Am. Soc. Nephrol. 2004, 15, 91–101.
- Chen, J.L.; Walton, K.L.; Winbanks, C.E.; Murphy, K.T.; Thomson, R.E.; Makanji, Y.; Qian, H.; Lynch, G.S.; Harrison, C.A.; Gregorevic, P. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014, 28, 1711–1723.
- Centrella, M.; McCarthy, T.L.; Canalis, E. Activin-A binding and biochemical effects in osteoblast-enriched cultures from fetal-rat parietal bone. Mol. Cell. Biol. 1991, 11, 250–258.
- Funaba, M.; Ogawa, K.; Abe, M. Expression and localization of activin receptors during endochondral bone development. Eur. J. Endocrinol. 2001, 144, 63–71.
- Solagna, F.; Patel, K.; Huber, T.B.; Solagna, F.; Tezze, C.; Lindenmeyer, M.T.; Lu, S.; Wu, G.; Liu, S.; Zhao, Y.; et al. Pro-cachectic factors link experimental and human chronic kidney disease to skeletal muscle wasting programs. J. Clin. Invesitig. 2021, 131, e135821.
- Lotinun, S.; Pearsall, R.S.; Davies, M.V.; Marvell, T.H.; Monnell, T.E.; Ucran, J.; Fajardo, R.J.; Kumar, R.; Underwood, K.W.; Seehra, J.; et al. A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone 2010, 46, 1082–1088.
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740.
- Arai, K.Y.; Ono, M.; Kudo, C.; Fujioka, A.; Okamura, R.; Nomura, Y.; Nishiyama, T. IL-1beta stimulates activin betaA mRNA expression in human skin fibroblasts through the MAPK pathways, the nuclear factor-kappaB pathway, and prostaglandin E2. Endocrinology 2011, 152, 3779–3790.
- Hully, J.R.; Chang, L.; Schwall, R.H.; Widmer, H.R.; Terrell, T.G.; Gillett, N.A. Induction of apoptosis in the murine liver with recombinant human activin A. Hepatology 1994, 20, 854–862.
- Maeshima, A.; Nojima, Y.; Kojima, I. Activin A: An autocrine regulator of cell growth and differentiation in renal proximal tubular cells. Kidney Int. 2002, 62, 446–454.
- Ishisaki, A.; Yamato, K.; Nakao, A.; Nonaka, K.; Ohguchi, M.; ten Dijke, P.; Nishihara, T. Smad7 is an activin-inducible inhibitor of activin-induced growth arrest and apoptosis in mouse B cells. J. Biol. Chem. 1998, 273, 24293–24296.
- Fukuchi, Y.; Kizaki, M.; Yamato, K.; Kawamura, C.; Umezawa, A.; Ji, H.; Nishihara, T.; Ikeda, Y. Mcl-1, an early-induction molecule, modulates activin A-induced apoptosis and differentiation of CML cells. Oncogene 2001, 20, 704–713.
- Liu, M.; Mao, C.; Li, J.; Han, F.; Yang, P. Effects of the Activin A-Follistatin System on Myocardial Cell Apoptosis through the Endoplasmic Reticulum Stress Pathway in Heart Failure. Int. J. Mol. Sci. 2017, 18, 374.
- Pasquale Esposito; Daniela Verzola; Daniela Picciotto; Leda Cipriani; Francesca Viazzi; Giacomo Garibotto; Myostatin/Activin-A Signaling in the Vessel Wall and Vascular Calcification. Cells 2021, 10, 2070, 10.3390/cells10082070.
More