Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Elisabetta Abruzzese.
Acute myeloid leukemia (AML), the most frequent acute leukemia in adults, has been historically treated with infusional cytarabine (ara-c) + daunorubicin (3 + 7) for at least 40 years. The first “target therapy” to be introduced was the monoclonal anti-CD33 gemtuzumab ozogamicin (GO) in 2004.
- acute myeloid leukemia
- gemtuzumab ozogamicin
- CD33
- chemotherapy
1. CD33 Expression in Normal Myelopoiesis
CD33, located on chromosome 19q13.3, is a sialic acid-binding immunoglobulin-related lectin (siglec) that functions as a transmembrane receptor on hematopoietic cells [1,2][1][2]. CD33 is comprised of an amino-terminal variable (V)-set Ig-like domain mediating sialic acid binding, a C2-set Ig-like domain in its extracellular region, a transmembrane domain, and a cytoplasmic tail that contains two conserved tyrosine-based inhibitory signaling motifs (ITIM), a property shared with all CD33-related siglecs discovered to date [3]. Alternative splicing of the CD33 RNA leads to a shorter isoform expressed on the cell surface. This isoform lacks the V-set Ig-like domain as well as the disulfide bond linking the V-and C2-set Ig-like domains [3]. It preferentially binds to α2-6-and α2-3-sialylated glycans and strongly binds to sialylated ligands on leukemic cell lines [4,5][4][5]. Downstream steps in CD33 signaling are not well known, but some experimental models report the involvement of several pathways, such as Syk, c-Cbl, Vav, and ZAP-70 [6,7][6][7]. CD33 works as an inhibitory receptor; when phosphorylated on its cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM) sequence, it recruits SHP-1 and SHP-2 phosphatases and downregulates cell activation, in both myeloid cell lines and activated natural killer (NK) cells [8].
Increasing evidence suggests that CD33 with inhibitory signaling motifs modulates inflammatory and immune responses through the dampening of tyrosine kinase-driven signaling pathways. In vitro studies have showed that CD33 constitutively inhibits the production of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-8 by human monocytes in a sialic acid ligand-dependent and SOCS3-dependent manner [6]. Conversely, reduction of cell surface CD33 expression or interruption of sialic acid binding can augment p38 mitogen-activated protein kinase (MAPK) activity and enhance cytokine secretion, as well as cytokine-induced cellular proliferation [6].
Physiologically, CD33 expression is restricted to early multi-lineage hematopoietic progenitors (CFU-GEMM, CFU-GM, CFU-G, and E-BFU), promyelocytes, myeloblasts and monoblasts, monocytes/macrophages, granulocyte precursors (with decreasing expression with maturation), and mast cells. On CD34+/CD33+ bone marrow cells, CD33 expression has been estimated to average around 8 × 103 molecules/cell, although levels vary widely (1–20 × 103 molecules/cell) [9]. Mature granulocytes may show a very low level of CD33 expression. In contrast, CD33 is not expressed outside the hematopoietic system nor is it expressed in pluripotent hematopoietic stem cells, erythrocytes, platelets, B, T, or NK cells [9].
2. CD33 Expression in Acute Myeloid Leukemia
The CD13 and CD33 antigens and myeloperoxidase that characterize normal myeloid progenitors are abundantly expressed on almost all acute myeloid leukemia (AML) cells [10]. Roughly, 85–90% of adult and pediatric AML are CD33-positive, as defined by the expression of this specific antigen on 20–25% or more of leukemic blasts. CD33 expression is essentially universal in acute promyelocytic leukemia (APL), with cells typically surrounded by large amounts (95–100%) of the antigen [11]. In addition, CD33 is also found on abnormal cells of other myeloid neoplasms (e.g., myelodysplastic syndromes and myeloproliferative neoplasms) [12], and on subsets of B-cell and T-cell acute lymphoblastic leukemia (ALL)/lymphoblastic lymphomas [13,14][13][14] perhaps consistent with its occasional expression on normal non-myeloid cells.
Bone marrow blasts of AML patients express between 709 and 54,894 CD33 molecules/cell (mean 10,380 molecules/cell), compared to only 859–5137 molecules/cell (mean 2997 molecules/cell) detected in normal CD33-positive bone marrow cells [12]. The CD33+/CD34+ AML subpopulations also express higher levels of CD33 antigen (mean 9482 molecules/cell) than normal bone marrow CD33+/CD34+ counterparts (mean 8154 molecules/cell) [12]. Furthermore, lower levels of CD33 have been detected on CD33+/CD34+ leukemic blasts (mean 7607 molecules/cell) of peripheral blood compared with bone marrow blasts, although most studies have not measured antigen levels on bone marrow and peripheral blood populations in individual patients [12]. CD33 expression in AML blasts using multi-parameter flow cytometry (MFC) is routinely measured both by median fluorescence intensity (CD33-MFI) and as a percentage of CD33 positivity. CD33-MFI is often detected in immunophenotypically immature CD34+CD38low stem/progenitor cells (SPC) [15]. A broad range of CD33-MFI and %CD33-positivity values are generally observed; therefore, in several studies, patients were grouped into increasing quartiles (Q1, Q2, Q3, Q4) according to CD33 blast expression levels [16,17,18][16][17][18].
Several studies have evaluated the correlation between CD33 expression and disease characteristics in AML patients [16,17,18][16][17][18]. In both adult and children, FLT3-ITD and NPM1 mutations were found to be significantly heightened in prevalence with increasing CD33 expression [13,16][13][16]. Moreover, intermediate-risk AML patients lacking these mutations were inversely associated with CD33 expression. In contrast, the significant increase in the prevalence of the protein CCAAT/enhancer-binding protein alpha (CEBPA) with increasing CD33 expression was not observed when analyzed by quartiles [16]. Core binding factor (CBF)-AML (t(8;21) (q22;q22) or inv(16) (p13q22)/t(16;16) (p13;q22) was also found to be inversely correlated with CD33 expression [16,18][16][18]. Khan et al. interestingly showed that the CD34+CD38- fraction of adult patients with CBF-AML expressed CD33, while patients with intermediate- and adverse-risk disease displayed a more heterogeneous CD34+CD38- fraction, containing significant numbers of CD33-negative cells [18]. This suggests that CBF-AML may have different patterns of CD33 expression, and removal of CD33+ leukemic stem cells (LSCs) may explain the favorable clinical response seen in adult patients receiving anti-CD33 therapy. Analysis from 1583 patients in the UK-NCRI-AML17 (younger adults) and UK-NCRI-AML16 (older adults) trials showed that cytogenetic adverse risk was associated with lower CD33 expression, while intermediate-risk cytogenetics significantly increased in prevalence with increasing CD33 quartiles [16]. In the pediatric AML population, there was an inverse association between CD33 expression and prevalence of low-risk AML; in contrast, the prevalence of standard-risk disease increased significantly with increasing quartile. There was no statistically significant trend in prevalence by quartile for high-risk disease [17]. Furthermore, pediatric trials showed that patients whose AML blasts display high CD33 levels experienced inferior disease-free periods and overall survival when treated with conventional chemotherapy that did not include CD33-targeted agents [17,19][17][19].
In AML patients, CD33 can also be detected as soluble protein in the circulation and may provide some prognostic information [20], however, its role as a predictive biomarker is still controversial. It is also unclear whether soluble CD33 might interfere with the therapeutic efficacy of CD33 antibodies, although some in vitro evidence suggests that soluble CD33 may not affect the activity of CD33-targeted immunotherapy [21].
3. Gemtuzumab Ozogamicin (GO) and Mechanism of Action
Gemtuzumab ozogamicin (GO) is a humanized murine IgG4 anti-CD33 antibody (P67.6) conjugated with a calicheamicin hydrazide derivative attached to the oxidized carbohydrates [22]. Unfortunately, 50% of the antibody remains unconjugated and the bare antibodies, like lintuzumab, retain limited clinical activity [23], yet, in theory, could deplete available CD33 on AML blasts without exerting cytotoxicity. CD33 is continually re-expressed on myeloid cells and returns to normal expression levels within 72 h from first GO infusion [24,25][24][25]. These data resulted in the advent of a fractioned schedule (days 1, 4, 7) at lower doses (3 mg/m2) than the first studies, which had the advantage of limiting hepatic toxicity (sinusoidal obstruction syndrome SOS or VOD). Mechanisms of SOS are not completely elucidated and are shared with other calicheamicin-bound antibody–drug conjugates (ADC) like inotuzumab ozogamicin (anti-CD22 antibody, approved for ALL) and vadastuximab talirine (SGN-CD33A with pyrrolobenzodiazepine as a different payload) [26]. Moreover, the parent antibody (P67.6) used for GO recognizes the V-set domain of CD33 and therefore interacts with CD33FL and CD33E7a but not with CD33ΔE2, nor with CD33ΔE2, E7a. In fact, a SNP placed in the splice enhancer region of the CD33 gene exon 2, rs12459419 (C > T; Ala14Val) is responsible for the lack of the V-set domain of CD33 and could impair the interaction of the drug with its receptor [27].
After GO binds to surface of CD33, the complex is internalized and transferred to the lysosomes where the acidic environment causes hydrolysis of the linker and the release of calicheamicin [28] (Figure 1). Nevertheless, efflux pump activity present in a large proportion of AML cells as the multidrug resistance-associated protein 1 (ABCC1 or MRP1) and MDR1 (or P-glycoprotein or ABCB1) can limit the effect of the free drug and accounts for the poor clinical outcome observed in studies [29]. This is also the case for GO, where higher expression of MDR1 is associated with poor outcome [30[30][31][32],31,32], and CD33 expression is inversely correlated with MDR1 drug efflux activity [3].
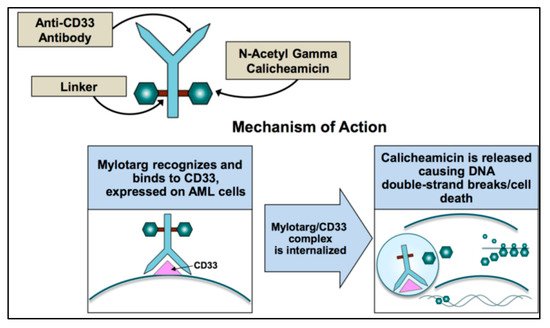
Figure 1. Mechanism of action of gemtuzumab ozogamicin.
Calicheamicin is a natural antibiotic of the nine-membered enediyne core family, with an anti-cancer effect 4000-fold more potent than adriamycin [33]. Calicheamicin produces a benzenoid diradical that, when positioned within the minor groove of DNA, extracts hydrogen atoms from the deoxyribose backbone of DNA. From its position on the DNA, the calicheamicin radical is able to cause inter-strand crosslinks, reacting with molecular oxygen to produce DNA double-strand breaks [34]. The extent of damage is related to the arrest of cell cycle in G2/M and impacts DNA repair mechanisms and apoptotic pathways. Cells with impaired mechanisms of repair (e.g., ATM, poly (ADP-ribose) polymerase-1 enzyme (PARP-1)) [35] are particularly sensitive to the cytotoxic effects of GO. If damage is overwhelming, cells will die through the mitochondrial pathway of apoptosis predominantly utilized for GO-induced cell death. The calicheamicin-ϑII derivative triggers apoptosis in a p53-independent and death receptor/FADD-independent manner via activation of the mitochondrial permeability transition, cytochrome-c release, and activation of caspase 9 and caspase 3 [36]. Therefore, the mitochondrial protein Bcl-2 together with other anti-apoptotic BCL-2 family members reduce the cytotoxicity of calicheamicin-GO [37,38][37][38].
Not surprisingly from the above discussion, in vitro samples of pediatric AML showed 100,000-fold difference in calicheamicin sensitivity between the most sensitive and the most resistant patient samples [39]. The reasons for this variability and resistance are still not completely elucidated.
References
- Freeman, S.; Kelm, S.; Barber, E.; Crocker, P. Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood 1995, 85, 2005–2012.
- Crocker, P.R. Siglecs: Sialic-acid-binding immunoglobulin-like lectins in cell–cell interactions and signalling. Curr. Opin. Struct. Biol. 2002, 12, 609–615.
- Walter, R.B.; Gooley, T.A.; Van Der Velden, V.H.J.; Loken, M.R.; Van Dongen, J.J.M.; Flowers, D.A.; Bernstein, I.D.; Appelbaum, F.R. CD33 expression and P-glycoprotein–mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 2007, 109, 4168–4170.
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850.
- Macauley, M.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666.
- Balaian, L.; Zhong, R.-K.; Ball, E.D. The inhibitory effect of anti-CD33 monoclonal antibodies on AML cell growth correlates with Syk and/or ZAP-70 expression. Exp. Hematol. 2003, 31, 363–371.
- Balaian, L.; Ball, E.D. Direct effect of bispecific anti-CD33 × anti-CD64 antibody on proliferation and signaling in myeloid cells. Leuk. Res. 2001, 25, 1115–1125.
- Hernández-Caselles, T.; Martínez-Esparza, M.; Pérez-Oliva, A.B.; Quintanilla-Cecconi, A.M.; García-Alonso, A.; Alvarez-López, D.M.R.; García-Peñarrubia, P. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: Two isoforms of CD33 are generated by alternative splicing. J. Leukoc. Biol. 2006, 79, 46–58.
- Andrews, R.; Torok-Storb, B.; Bernstein, I. Myeloid-associated differentiation antigens on stem cells and their progeny identified by monoclonal antibodies. Blood 1983, 62, 124–132.
- Brendel, C.; Neubauer, A. Characteristics and analysis of normal and leukemic stem cells: Current concepts and future directions. Leukemia 2000, 14, 1711–1717.
- Guglielmi, C.; Martelli, M.P.; Diverio, D.; Fenu, S.; Vegna, M.L.; Cantù-Rajnoldi, A.; Biondi, A.; Cocito, M.G.; Del Vecchio, L.; Tabilio, A.; et al. Immunophenotype of adult and childhood acute promyelocytic leukaemia: Correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br. J. Haematol. 1998, 102, 1035–1041.
- Jilani, I.; Estey, E.; Huh, Y.; Joe, Y.; Manshouri, T.; Yared, M.; Giles, F.; Kantarjian, H.; Cortes, J.; Thomas, D.; et al. Differences in CD33 Intensity Between Various Myeloid Neoplasms. Am. J. Clin. Pathol. 2002, 118, 560–566.
- Handgretinger, R.; Schäfer, H.-J.; Baur, F.; Frank, D.; Ottenlinger, C.; Bühring, H.-J.; Niethammer, D. Expression of an early myelopoietic antigen (CD33) on a subset of human umbilical cord blood-derived natural killer cells. Immunol. Lett. 1993, 37, 223–228.
- Raponi, S.; De Propris, M.S.; Intoppa, S.; Milani, M.L.; Vitale, A.; Elia, L.; Perbellini, O.; Pizzolo, G.; Foa, R.; Guarini, A. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: Analysis of 552 cases. Leuk. Lymphoma 2011, 52, 1098–1107.
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208.
- Pollard, J.A.; Alonzo, T.A.; Loken, M.; Gerbing, R.B.; Ho, P.A.; Bernstein, I.D.; Raimondi, S.C.; Hirsch, B.; Franklin, J.; Walter, R.B.; et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood 2012, 119, 3705–3711.
- Olombel, G.; Guerin, E.; Guy, J.; Perrot, J.-Y.; Dumézy, F.; De Labarthe, A.; Bastie, J.-N.; Legrand, O.; Raffoux, E.; Plesa, A.; et al. The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood 2016, 127, 2157–2160.
- Khan, N.; Hills, R.; Virgo, P.; Couzens, S.; Clark, N.; Gilkes, A.; Richardson, P.; Knapper, S.; Grimwade, D.; Russell, N.H.; et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia 2017, 31, 1059–1068.
- Dinndorf, P.A.; Buckley, J.D.; Nesbit, M.E.; Lampkin, B.C.; Piomelli, S.; Feig, S.A.; Kersey, J.H.; Hammond, G.D.; Bernstein, I.D. Expression of myeloid differentiation antigens in acute nonlymphocytic leukemia: Increased concentration of CD33 antigen predicts poor outcome—A report from the childrens cancer study group. Med. Pediatr. Oncol. 1992, 20, 192–200.
- Biedermann, B.; Gil, D.; Bowen, D.T.; Crocker, P.R. Analysis of the CD33-related siglec family reveals that Siglec-9 is an endocytic receptor expressed on subsets of acute myeloid leukemia cells and absent from normal hematopoietic progenitors. Leuk. Res. 2020, 31, 211–220.
- Friedrich, M.; Henn, A.; Raum, T.; Bajtus, M.; Matthes, K.; Hendrich, L.; Wahl, J.; Hoffmann, P.; Kischel, R.; Kvesic, M.; et al. Preclinical Characterization of AMG 330, a CD3/CD33-Bispecific T-Cell–Engaging Antibody with Potential for Treatment of Acute Myelogenous Leukemia. Mol. Cancer Ther. 2014, 13, 1549–1557.
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjugate. Chem. 2002, 13, 47–58.
- Sekeres, M.A.; Lancet, J.E.; Wood, B.L.; Grove, L.E.; Sandalic, L.; Sievers, E.; Jurcic, J.G. Randomized, phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica 2012, 98, 119–128.
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153.
- Van Der Velden, V.H.J.; Te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204.
- Godwin, C.D.; McDonald, G.B.; Walter, R.B. Sinusoidal obstruction syndrome following CD33-targeted therapy in acute myeloid leukemia. Blood 2017, 129, 2330–2332.
- Lamba, J.K.; Pounds, S.; Cao, X.; Downing, J.R.; Campana, D.; Ribeiro, R.C.; Pui, C.H.; Rubnitz, J.E. Coding polymorphisms in CD33 and response to gemtuzumab ozogamicin in pediatric patients with AML: A pilot study. Leukemia 2009, 23, 402–404.
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody—Calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjug. Chem. 2002, 13, 40–46.
- Boyer, T.; Gonzales, F.; Barthélémy, A.; Marceau-Renaut, A.; Peyrouze, P.; Guihard, S.; Lepelley, P.; Plesa, A.; Nibourel, O.; Delattre, C.; et al. Clinical Significance of ABCB1 in Acute Myeloid Leukemia: A Comprehensive Study. Cancers 2019, 11, 1323.
- Larson, R.A.; Sievers, E.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452.
- Sievers, E.; Larson, R.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and Safety of Gemtuzumab Ozogamicin in Patients With CD33-Positive Acute Myeloid Leukemia in First Relapse. J. Clin. Oncol. 2001, 19, 3244–3254.
- Linenberger, M.L.; Hong, T.; Flowers, D.; Sievers, E.L.; Gooley, T.A.; Bennett, J.M.; Berger, M.S.; Leopold, L.H.; Appelbaum, F.R.; Bernstein, I.D. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001, 98, 988–994.
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sepehr, K.S.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64.
- Kennedy, D.R.; Ju, J.; Shen, B.; Beerman, T.A. Designer enediynes generate DNA breaks, interstrand cross-links, or both, with concomitant changes in the regulation of DNA damage responses. Proc. Natl. Acad. Sci. USA 2007, 104, 17632–17637.
- Sullivan, N.; Lyne, L. Sensitivity of fibroblasts derived from ataxia-telangiectasia patients to calicheamicin γ1I. Mutat. Res. Lett. 1990, 245, 171–175.
- Prokop, A.; Wrasidlo, W.; Lode, H.; Lang, F.; Henze, H.; Dörken, B.; Wieder, T.; Daniel, P.T. Induction of apoptosis by enediyne antibiotic calicheamicin θII proceeds through a caspase-mediated mitochondrial amplification loop in an entirely Bax-dependent manner. Oncogene 2003, 22, 9107–9120.
- Mei, M.; Aldoss, I.; Marcucci, G.; Pullarkat, V. Hypomethylating agents in combination with venetoclax for acute myeloid leukemia: Update on clinical trial data and practical considerations for use. Am. J. Hematol. 2018, 94, 358–362.
- Godwin, C.D.; Bates, O.M.; Jean, S.R.; Laszlo, G.S.; Garling, E.E.; Beddoe, M.E.; Cardone, M.H.; Walter, R.B. Anti-apoptotic BCL-2 family proteins confer resistance to calicheamicin-based antibody-drug conjugate therapy of acute leukemia. Leuk. Lymphoma 2020, 61, 2990–2994.
- Goemans, B.F.; Zwaan, C.M.; Vijverberg, S.J.H.; Loonen, A.H.; Creutzig, U.; Hählen, K.; Reinhardt, D.; Gibson, B.E.S.; Cloos, J.; Kaspers, G.J.L. Large interindividual differences in cellular sensitivity to calicheamicin may influence gemtuzumab ozogamicin response in acute myeloid leukemia. Leukemia 2008, 22, 2284–2285.
More