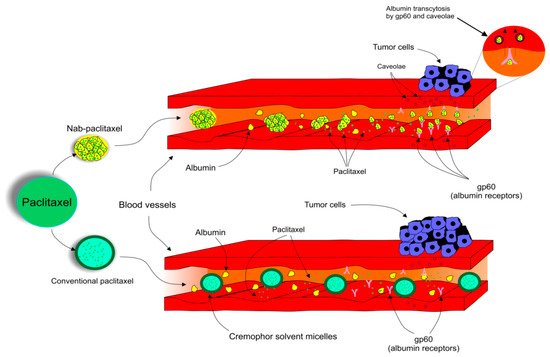
Paclitaxel (PTX), the most widely used anticancer drug, is applied for the treatment of various types of malignant diseases. Mechanisms of PTX action represent several ways in which PTX affects cellular processes resulting in programmed cell death. PTX is frequently used as the first-line treatment drug in breast cancer (BC). Unfortunately, the resistance of BC to PTX treatment is a great obstacle in clinical applications and one of the major causes of death associated with treatment failure. Factors contributing to PTX resistance, such as ABC transporters, microRNAs (miRNAs), or mutations in certain genes, along with side effects of PTX including peripheral neuropathy or hypersensitivity associated with the vehicle used to overcome its poor solubility, are responsible for intensive research concerning the use of PTX in preclinical and clinical studies. Novelties such as albumin-bound PTX (nab-PTX) demonstrate a progressive approach leading to higher efficiency and decreased risk of side effects after drug administration. Moreover, PTX nanoparticles for targeted treatment of BC promise a stable and efficient therapeutic intervention.
1. Introduction
A crucial aspect of the modern era is the rapid progress in the prevalence of many civilization diseases including cancer
[1]. Breast cancer (BC )is the most commonly occurring malignant disease in women and the leading cause of cancer death among them and still remains a global problem of public health
[2]. Modern approaches in the field of oncology aimed at BC including novelties in diagnosis, treatment, and prevention have a crucial role in the management of cancer. Better knowledge of the biologic heterogeneity of BC leads to the development of more effective therapy concepts in personalized medicine
[3]. Over the last decades, substantial progress in the treatment of BC led to the discovery of new drugs with specific actions in cancer suppression. Currently, there are several classes of chemo-therapeuticals based on antimetabolites, alkylating agents, immunological elements, hormonal components, or mitotic deprivation
[4]. Recently, two groups of chemotherapeutic drugs (anthracyclines and taxanes) were widely used in adjuvant and neoadjuvant treatment of BC
[5]. Paclitaxel (PTX), a class of taxanes, is an antineoplastic drug with an impact on the stabilization of microtubules, which represents a widely used chemotherapeutic agent in numerous cancers. The effect of PTX as an antimitotic drug was documented in a large number of studies
[6][7][8][6,7,8]. Moreover, the mechanisms of PTX action associated with the inhibition of tumor growth can act on different levels. In these studies, PTX initiated a cascade of signaling pathways resulting in programmed cell death
[9][10][9,10]. Modulation of epigenetic markers represents the novelty of cancer-related research, and PTX may also regulate the expression of certain microRNAs (miRNAs) associated with cancer progression. Furthermore, PTX can exert a variety of positive influences on the modulation of immune response via regulation of chemokines, cytokines, or immune cells
[11][12][11,12]. The resistance of BC to PTX and other chemotherapeutics as a consequence of disequilibrium in various signaling pathways, mutations in certain genes, and epigenetic deregulations is responsible for the worse clinical outcome for patients with BC
[13][14][15][13,14,15]. The global challenge in the application of PTX as a dominant anticancer chemotherapeutic agent is the reduction of side effects and increasing drug efficiency. Novelties such as Albumin-bound PTX(nab-PTX) are awesome examples of the progress in the oncology-associated area focused on cross-connection of nanotechnology and cancer treatment
[16].
2. Paclitaxel: Fundamental Drug in Chemotherapy and Novel Advances in its Application
As noted above, antimitotic chemotherapeutics suppress the polymerization dynamic of microtubules, resulting in the induction of mitotic arrests as a consequence of the activation of the mitotic checkpoint. Consequently, PTX, a member of taxanes, represents one of the most important antineoplastic drugs frequently used in the treatment of numerous types of cancers including BC.
2.1. The Origin of Paclitaxel
PTX, trademarked by Bristol-Myers Squibb (BMS) as Taxol™ in 1992
[17][57], is an antimitotic, anticancer drug that was approved by the FDA (Food and Drug Administration) in 1994 for use in BC
[18][58]. PTX is clinically used to treat solid tumors such as ovarian cancer, hormone refractory prostate cancer, and non-small cell lung cancer
[19][59]. The active ingredient was first isolated from the Pacific Yew tree (
Taxus brevifolia) by Mansukh Wani and Monroe Wall
[20][60]. The drug went through additional clinical trials and testing on mouse tumor models before it was FDA approved for ovarian cancer in 1992
[17][57]. Due to the high demand for the drug, the slow-growing
T. brevifolia tree was not able to provide the market and research’s needs; therefore, it was concluded that the production of PTX from
T. brevifolia was impractical, non-environmentally conscious, and financially burdening according to the National Cancer Institute (NCI)
[21][22][61,62]. In 1993, a new method of mass-producing PTX was associated with a fungus isolated from the phloem of
T. brevifolia [23][63]. In 1994, a successful semi-synthetic approach of synthesizing PTX was formulated and approved by the FDA, which is the method of production until today
[18][22][58,62].
2.2. Paclitaxel’s Mechanism of Action
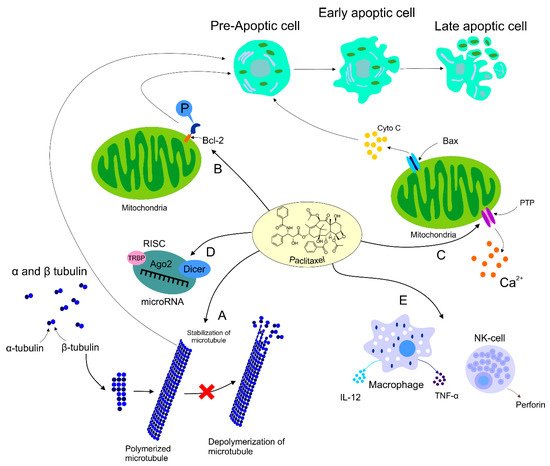
(I) Paclitaxel as a Polymerization Factor
PTX binds to microtubules instead of tubulin dimers and stabilizes microtubules (polymerization) by promoting the assembly of alpha and beta tubulin subunits, the building blocks of microtubules
[17][24][57,64]. The drug reduces the critical concentration of tubulin required for its assembly, therefore promoting the lengthening of the tubulin polymer
[25][65]. The stability of the microtubules interferes with microtubules´ dynamics. Subsequently, the cell’s ability to divide is disrupted due to insufficient requirements of the mitotic checkpoint; therefore, cell division halts at the G2 or M phase. The polymerized and stable microtubules remain largely unaffected even by cold temperatures and calcium. The presence of calcium reduces PTX’s affinity for tubulin; therefore, the equilibrium of polymerization/depolymerization shifts towards polymerization to offset this effect
[17][26][27][57,66,67]. Moreover, chondrocytes show that PTX causes cytoskeletal abnormalities in which microtubules become stubby and straight in the cytoplasm, with rough endoplasmic reticulum as opposed to fine, sinous filaments in the control group. These changes persist for 48 h after the removal of PTX. The changed microtubules dislodge ribosomes off the rough endoplasmic reticulum and fuse nearby endoplasmic reticulum complexes together
[26][66]. PTX polymerizes only free microtubules not attached to or preexisting in the microtubule organizing centers (MTOC). Attached microtubules disappear in the presence of PTX
[28][68]. PTX interferes with the dynamics of microtubules and microtubule polymerization and delays the progression of mitosis by inducing failure in chromosomal segregation, all of which eventually lead to the induction of apoptosis and mitotic arrest
[29][30][31][69,70,71].
(II) Paclitaxel’s Effect Depends on Concentration
The mechanism of PTX cytotoxicity highly depends on the concentration of the drug in the cell as demonstrated in in vitro studies. Giannakakou et al.
[32][72] documented that the reduction of proliferation of the lung carcinoma cell line A549 as well as breast MCF-7 cells after treatment by PTX at concentrations above 12 nM resulted in G2/M arrest. Interestingly, lower concentrations of PTX (3–6 nM) exerted similar potential to suppress the proliferation of cancer cells, resulting in programmed cell death
[32][72]. A study focusing on drug concentration analyzed the effect of low doses of PTX (10 nM) on cancer cell invasiveness. In this in vitro study, researchers evaluated the impact of the non-anti-mitotic concentration of PTX resulting in the reduction of transwell invasion of MDA-MB-231 as a consequence of the regulation of voltage-dependent sodium channel expression
[33][73]. Additionally, the potential role of low doses of PTX (20 nM) combined with a Wnt signaling inhibitor regulated the molecular events, including E-cadherin upregulation and β-catenin reduction, leading to suppression of tumor growth, metastasis, and angiogenesis in BC
[34][74].
(III) Paclitaxel Affects Phosphorylation of Bcl-2
Although many researchers agree that the cytotoxicity of PTX lies in its ability to cause Bcl-2 hyperphosphorylation, many other studies report dephosphorylation of Bcl-2 coinciding with apoptosis
[35][36][37][75,76,77]. However, since apoptosis does not occur immediately after exposure to PTX, the duration of exposure and constant Bcl-2 phosphorylation contribute to the drug’s cytotoxicity
[38][78]. On the other hand, different studies proved that phosphorylated Bcl-2 does not dimerize with BAX; therefore, it is argued that the unassociated BAX favors apoptosis
[39][40][79,80]. However, the later studies are older and probably less updated than the former studies, and these conclusions are based on prostate and leukemia cell lines.
(IV) Paclitaxel Affects Calcium Signaling
PTX induces the depletion of calcium ions from the mitochondrial reserve through the mitochondrial permeability transition pore (PTP). The leaving calcium induces PTP to release the apoptogenic factor cytochrome C (cyto C) into the cytosol from the mitochondria to initiate apoptosis
[41][42][43][81,82,83]. It is believed that side effects of antimitotic drugs are related to the role of those drugs in the calcium signal cascade. The severity and heterogeneity of these side effects are attributed to the alterations in mitochondrial calcium uptake in different cells. Using extremely high doses of PTX results in the rupture of the mitochondria, the release of cyto C, and the initiation of apoptosis without the efflux of calcium
[44][84]. Reasons why peripheral neurons are primarily affected by PTX are not known, even though most cells have mitochondria. On the other hand, PTX induces apoptosis in the presence of an extracellular calcium reservoir via calcium influx when the drug is administered in high doses. However, low doses of PTX demonstrated patterns of apoptosis independent of the extracellular concentration of calcium
[10].
(V) The impact of Paclitaxel on microRNA Expression Profiles
MiRNA, small non-coding RNA with a regulatory function in gene expression, can be regulated by various antineoplastic drugs including PTX. Several studies focusing on miRNA expression demonstrated cross-connection between an application of the drug and alterations in miRNA expression profiles. After PTX intervention, expression levels of let-7a and miR-205 with tumor suppressor potential targeting K-Ras and HER3 were changed in the BC cell line BT-474
[45][85]. Interestingly, metronomic treatment (low dose LDM) by PTX reduced the level of let-7f, while the expression of thrombospondin-1 (TSP-1) associated with anti-angiogenic potential was increased in PTX LDM therapy
[46][86]. In summary, preclinical trials demonstrated the modulatory potential of PTX in the regulation of miRNA expression, but further research is needed for a better implication of the drug in BC treatment.
(VI) The Immunomodulatory Effects of Paclitaxel
The role of PTX was also documented in the area of immunomodulation, with both stimulation and suppression of immune cells associated with tumor growth. On the other hand, the suppression of immune cells could have a negative impact on the host immune response against cancer development
[47][87]. Increasing evidence supports participation of PTX in the regulation of host immunity via stimulation of macrophages leading to cytokine secretion including TNF-α or IL-12 that induce activation of natural killers (NK), dendric cells (DC), and cytotoxic T lymphocytes resulting in the eradication of tumor cells
[11][48][11,88]. Additionally, direct-acting PTX was evaluated in DC via binding to Toll-like receptor localized on the DC surface, thus promoting maturation of antigen-presenting cells
[49][89]. Furthermore, the dose-dependent administration of PTX led to an increased level of MHC class II
[50][90]. The impact of PTX on immunomodulation was also identified in NK cells. A higher level of cytotoxicity correlated with an increased level of perforin representing the crucial effector protein of NK activity, resulting in the premise that PTX enhanced NK cytotoxicity in a dose-dependent manner
[51][91]. Further research focusing on the regulation of the immune response in cancer is necessary for better understanding of an association between effects of PTX and immunopharmacology. The mechanisms of PTX in antineoplastic processes described above are summarized in
Figure 1.
Figure 1. Mechanism of action of PTX. Anti-tumor mechanism of action of PTX leading to stabilization of microtubule, cell arrest, and subsequent apoptosis (A). PTX also causes activation of the immune response contributing to tumor eradication (B). The ability of PTX to inactivate Bcl-2 via phosphorylation of the anti-apoptotic protein resulting in apoptosis (C). Participation of PTX in the regulation of certain miRNAs associated with the modulation of tumor progression (D). Regulation of calcium signaling by PTX results in PTX-induced release of cyto C from the mitochondria and programmed cell death.
2.3. Paclitaxel’s Effect on HER2+ Breast Cancer
The HER2+ subset of BC is more actively biochemically studied as opposed to other subsets. The efficacy of PTX in HER2+ BC patients was inconsistent and contradicting. In 1998, HER2+ BC was found to be biochemically resistant to PTX. The over-expression of HER2 upregulates p21cip1, which inhibits p34cdc2 that is normally activated by PTX in order to induce apoptosis of cancerous cells at the G2/M phase; therefore, the cytotoxic effect of PTX was inhibited
[52][92]. More recent studies conducted on 3121 node-positive postoperative patients showed that the addition of PTX in adjunction to doxorubicin plus cyclophosphamide decreased the rate of recurrence and death significantly upon 10-year follow up
[53][93]. In contrast, out of 46.7% patients responding to taxanes, 65.2% represented HER2+ and 35.5% HER2− tumors
[54][94]. The author suggested that PTX works on a signaling transduction pathway specific to HER2+ cancer. PTX activates the tumor suppressant protein p35 and the CDK inhibitor p21WAF1. On the other hand, p21WAF1 is not activated due to the usual activation of p35 since cancer cells lack the presence of this tumor suppressor. Nevertheless, it is suggested that p21WAF1 activation is c-raf-1 dependent since PTX activates c-raf-1, and the accumulation of p21WAF1would not be possible in the case of c-raf-1 depletion
[54][55][94,95].
2.4. Dose Ranges Administered
A number of studies evaluated different doses of PTX
[56][96].
Table 2 demonstrates the established dose ranges clinically administered to BC patients according to the state and diagnosis of the patient.
Table 2. The administered amount of PTX in correspondence with the patient’s condition and diagnosis.
| Condition |
Administration Schedule |
Concentration Range |
Reference |
| Adjuvant therapy with doxorubicin (node-positive or high-risk node-negative BC) |
Every 3 weeks |
175 mg/m2 IV perfusion over 3 h (4 courses) |
[57][97] |
| Weekly |
80 mg/m2 IV perfusion over 1 h (12 courses) |
[58][98] |
| Failure of neoadjuvant therapy (MBC or relapse within 6 months of neoadjuvant therapy) |
Every 3 weeks |
175 mg/m2 IV perfusion over 3 h |
[57][97] |
| Untreated MBC |
Every 3 weeks (max. of 8 cycles) |
200 mg/m2 IV infusion over 3 h + total dose of 480 mg/m2 doxorubicin
25 mg oral prednisone pre-treatment (12 h before treatment)
10 mg intramuscular chlorpheniramine + 300 mg intravenous cimetidine (both 30 min before PTX) |
[59][99] |