Arginine is an amino acid critically involved in multiple cellular processes including the syntheses of nitric oxide and polyamine, and is a direct activator of mTOR, a nutrient sensing kinase strongly implicated in carcinogenesis. In this review, we will discuss arginine as a signaling metabolite, arginine’s role in cancer metabolism, arginine as an epigenetic regulator, arginine as an immunomodulator, and arginine as a therapeutic target. The different cell killing mechanisms associated with various cancer types will also be described.
- arginine
- cancer metabolism
- epigenetics
- arginine-deprivation therapy
- ADI
- arginase
1. Introduction
An important step in tumor development is a metabolic adaptation to cope with the demand of rapid cell division as well as a hypoxia, and nutritionally deprived microenvironment[1]. Different tumors utilize different strategies to reprogram their metabolic pathways. In so doing, tumor cells expose specific vulnerabilities, which can be exploited therapeutically. For instance, tumor cells, not their normal counterparts, are “addicted” to certain external nutrients including amino acids and amino acid starvation therapy has gained significant momentum in recent years[2]. One of the most common metabolic defects of tumor cells is the impaired intrinsic ability to synthesize arginine[3]. Targeting exogenous arginine by arginine-metabolizing enzymes such as arginase, arginine decarboxylase and arginine deiminase (ADI) has received increasing attention as therapies to treat a variety of cancers[4]. There are a number of excellent reviews on this topic[5][6][7]. In this review, we will focus on recent progress in understanding arginine’s role in cancer metabolism as a signaling metabolite, an epigenetic regulator and an immunomodulator. As much of the knowledge was derived from characterizing arginine-deprived cancer cells, we will also update the current status of arginine-deprivation therapy.
2. Arginine and Signal Transduction
There are at least two ways arginine can transmit signals to the cells. The first is through transporters, solute carriers (SLCs) (Figure 1). As a cationic amino acid, arginine is mainly imported by two types of SLCs, the cationic amino acid transporters and the system y + L amino acid transporters[8][9]. It is noteworthy that arginine activates its downstream mTOR signal via lysosomal SLC38A9[10]. Arginine is the most consumed amino acid in the inner necrotic core of tumor mass, indicating its high demand for the survival of tumor cells[11]. Accordingly, tumor cells frequently overexpress specific types of SLCs such as SLC6A14, SLC7A3, SLC7A9, etc. to meet their high arginine demand. It should be noted that T cells up-regulate distinct types of SLCs to increase arginine uptake for T cell activation and anti-tumor functions[12][13]. Thus, targeting the tumor specifically with SCLs, and avoiding those expressed in T-cell and macrophage (e.g., SLC7A1, and A2) could be a potential strategy for cancer therapy.
Subsequent to its transport, arginine is able to activate several signal pathways. Chief among them is mTOR kinase. Arginine is one of only three amino acids that can directly activate mTOR pathway, a major cellular sensor of nutritional state[14]. The other two are glutamine and leucine. As such, arginine has profound impacts on protein synthesis, lipid synthesis and nucleotide synthesis, three anabolic pathways mediated by mTOR[15]. Indeed, nutrients[16][17]. are as important as growth factors in the activation of mTOR. Upon growth factor stimulation, mTOR can be activated through either PI3K (phosphatylinositol 3-kinase) pathway or MAPK pathway, via the inactivation of TSC (tuberous sclerosis complex), an mTOR negative regulator[18]. Inhibition of TSC converts the Rheb (RAS homolog enriched in brain) into active form, resulting in the activation of mTORC1 (mTOR complex 1). There are at least three ways, arginine can activate mTOR. (1) arginine disrupts the interaction between TSC and mTORC1, thereby activating mTOR[19]. (2) in the lysosome, arginine interacts with SLC38A9 and v-ATPase, upstream regulators of mTORC1, leading to the activation of Rag GTPase that is required for recruitment of mTORC1 complex to the lysosomal surface[20][21]. (3) in the cytosol, arginine interacts with CASTOR1(cytosolic arginine sensor for mTORC1 subunit 1) to disrupt the CASTOR complex, which is a negative regulator of Rag A[22], allowing Rag A to bind mTORC1 component RAPTOR (Regulatory-associated protein of mTOR) and redistributes mTORC1 to the lysosome. This may explain why arginine is such a potent activator of mTOR, and arginine deprivation leads to immediate inactivation of mTOR.
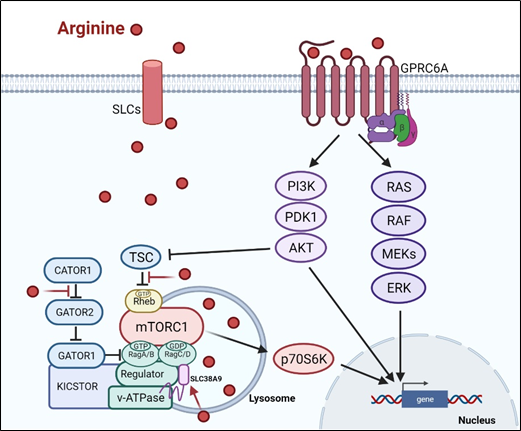
Figure 1. Arginine-related signaling pathway
Arginine-related signaling pathway
3. Arginine and Epigenetic Regulation
Recent studies showed that arginine can act as an effective epigenetic modulator[23]. In cancer cells, arginine is a strong inducer of histone acetylation, globally enhancing the expression of metabolic, mitochondrial and DNA repair genes. Histone acetylation involves the transfer of acetyl group from acetyl-CoA to histone mediated by HATs (histone acetyltransferases) and KATs (lysine acetyltransferases), which is counteracted by deacetylation enzymes such as HDACs (histone deacetylases) and SIRTs (sirtuins). Several enzymes including ACLY (ATP citrate synthase), ACSS1 (Acyl-coA synthetase short-chain family member 1) and ACSS2 (Acyl-coA synthetase short-chain family member 2) contribute to the synthesis of acetyl-CoA. In arginine stimulated cells, the acetyl-CoA level significantly increases so do the expression levels of ACLY, ACSS2 and the majority of HATs and KATs. By contrast, the expressions of several of the HDACs and SIRTs are decreased. These results together could account for the increased global histone acetylation observed. Since mTOR is known to activate ACLY and ACSS[24][25], arginine stimulation of histone acetylation is in part attributed to the activation of mTOR. The global increase of histone acetylation however is not random but has region specificity, which is dictated by several transcription factors including TEAD4, STAT3 (signal transducer and activator of transcription 3), WT1 (Wilms’ tumor 1) and TFAM ( mitochondrial transcription factor A).
Conversely, arginine deprivation leads to depletion of α-KG, which has profound effects on epigenetic regulation. As described above, arginine deprivation immediately affects mitochondrial functions[26][27][28][29] with consequent depletion of mitochondrial metabolites including α-KG. Alpha-KG is a cofactor of jumonji domain C containing histone demethylases (KDMs). As such, histone methylation generally increases during arginine deprivation. Most prominent are H3K9me3 and H3K27me3, two repressive marks contributing to gene silencing. These marks decorate genes involved in mitochondrial functions including OXPHOS, purine and pyrimidine synthesis, DNA repair, etc. (Figure 2). The consequence of such epigenetic repression is mitochondrial dysfunction, generation of reactive oxygen species (ROS), DNA damage and slow DNA repair, features which figure prominently in arginine-deprived tumor cells[26][27][28].
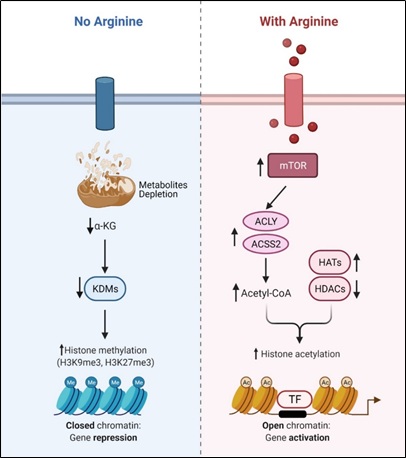
Figure 2. Arginine acts as Arginine acts as an epigenetic regulator.
an epigenetic regulator
4. Arginine and Genome Integrity
Sufficient arginine is required for maintaining nucleotide pool and DNA repair capacity. Although arginine is not directly involved in the synthesis of nucleotide, arginine can be converted to glutamine, proline and serine, precursors of pyrimidine and arginine abundance affects genome integrity. As described above, arginine augments the transcription of genes involved in purine and pyrimidine synthesis. In addition, by virtue of activating mTOR/S6K pathway, arginine promotes the phosphorylation and oligomerization of CAD complex (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase) to enhance pyrimidine synthesis[30][31]. On the other hand, both arginine and pyrimidine syntheses require aspartate and they “compete” for this metabolite. Tumor cells often augment pyrimidine synthesis by suppressing arginine synthesis via epigenetic silencing of ASS1[32][33], the basis of arginine-deprivation therapy. As described above, arginine deprivation also activates ATF4/ASAN which converts aspartate to asparagine causing depletion of nucleotide pool. In this scenario, the cell death caused by arginine-deprivation can be partially rescued by the addition of aspartate or nucleotide precursors [27].
In addition to arginine’s ability to epigenetically regulate the transcription of DNA repair genes, arginine affects DNA repair through the synthesis of polyamines. Polyamines interact with negatively charged DNA and plays a key role in maintaining the genome stability [34]. Polyamine depletion impairs DNA repair[35] and sensitizes cancer cells to genotoxic reagents[36][37]. Consistently, arginine deprivation which significantly reduces the polyamine levels[38] synergizes with polyamine inhibitors in the killing of cancer cells[38].
As arginine deficiency both depletes nucleotide pool and slows down DNA repair in tumor cells, it is no surprise that arginine starved tumor cells exhibit extensive DNA damages[39][27][28]. In ASS1-low pancreatic ductal adenocarcinoma, arginine deprivation exacerbates the HDAC inhibition-induced downregulation of C-terminal-binding protein-interacting protein (CtIP), a key protein for homologous recombination, leading to DNA damage and cell death[33]. In prostate and pancreatic cancer cells, arginine-starvation induced caspase-independent autophagic cell death with the appearance of nuclear DNA leakage and chromatin-autophagy (chromatophagy)[39]. This is caused by mitochondrial dysfunction and ROS production in the presence of excessive autophagy. Depletion of mitochondria or removal of ROS by NAC attenuates the DNA leakage phenotype and cell death[27]. In a study of ASS1-low melanomas, regardless of the BRAF status, arginine deprivation down-modulates FANCD2 and p-ATM, which are important initiators for DNA double strand break repair[40]. Although in this cell type, arginine deprivation alone does not induce DNA damage, combined treatment with cisplatin increases DNA double breaks, possibly due to persistent downregulation of DNA repair machinery caused by arginine deprivation. Taken together, arginine affects nucleotide synthesis/DNA repair in a complex way. The nucleotide insufficiency and down-modulated DNA repair machinery may underlie the arginine deprivation-induced DNA damage and its deficiency impairs this process and causes death of tumor cells.
5. Arginine and Immunomodulation
Arginine is a crucial immune-modulating amino acid for both innate and adaptive immunity[41][42]. It is involved in the activation of T-cell via the upregulation of T-cell receptor[43] and accelerating cell cycle progression[13]. Depletion of arginine has been used by tumor cells to generate an immunosuppressive micro-environment. Cancer cells release factors (G-CSF, GM-CSF, CCl2, etc.) to convert myeloid cells into immunosuppressive phenotypes (e.g., MDSC, myeloid-derived suppressive cells, or M2 macrophages)[44].
Although the correlation between arginine removal by arginase and T cell suppression has been well established, how systemic removal of arginine affects the tumor microenvironment and tumor growth remains poorly understood. In a study of the effect of autophagy on tumor growth, it was found that autophagy-negative (Atg) mouse released abundant arginase in the serum with consequent systemic depletion of arginine[45]. In these mice, ASS1-low syngeneic murine melanoma cells failed to grow with the infiltration of CD8 positive cells, indicating the T cell immune response is not severely affected by systemic depletion of arginine.
In another study, when human peripheral blood mononuclear cells (PBMCs) are stimulated by anti-CD3/CD28 antibodies, co-treatment with ADI-PEG20 does not block T cell activation; instead, arginine deprivation sustains the CD69+ T cells up to 72 h[46]. In the meantime, the induction of CTLA4 and PD-1 in activated T cells is blunted by arginine deprivation, and arginine deprivation also prevents Treg cells differentiation. Although arginine deprivation decreases cell proliferation of activated T cell as shown by previous studies, T cell infiltration is not compromised in syngeneic models[45][46].
Increasing evidence shows that arginine deprivation induces autonomous cancer cell death and enhances immune response. Dietary arginine-restriction offers a promising option for prevention and intervention.
6. Arginine Deprivation and Cell Killing
Arginine deprivation suppresses the growth and induces cell death of ASS1-low cancer cells. The general mechanisms associated with cell killing have been studied in a number of systems (Figure 3).
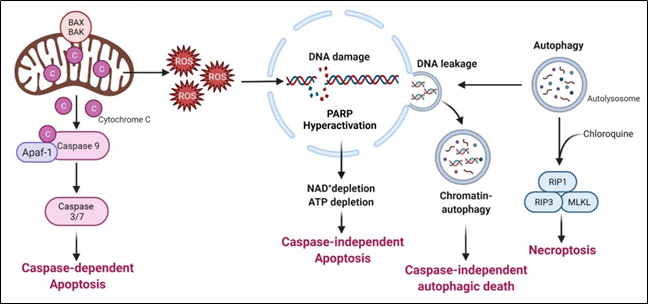
Figure 3. Arginine deprivation-induced types of cell death.
Arginine deprivation-induced types of cell death
6.1. Caspase-Dependent Apoptosis
This is the major mechanism associated with arginine-deprivation induced cell death, which operates in many cancer types including pleural mesothelioma cells[47], lymphoma cells[48], pancreatic cancer cells[33][49], ovarian cancers[50], sarcoma cells[51], T-lymphoblastic leukemia cells[52], liver cancer cells[53] and melanoma[54].
6.2. Caspase-Independent Apoptosis
Syed N et al. and Kelly MP reported that in some glioma cells and small cell lung carcinoma respectively, arginine deprivation-induced apoptosis, but it is caspase-independent[55][56]. The detailed mechanism remains to be elucidated.
6.3. Caspase-Independent Autophagic Death
Arginine deprivation inhibits mTOR, which is a negative regulator of autophagy. Accordingly, arginine deprivation is often accompanied by aggressive autophagy. Autophagy is a major means to regenerate arginine, which protects cells from nutrient stress. However, prolonged arginine deprivation leads to excessive and aberrant autophagy. This, coupled with ROS-induced DNA damage, leads to chromatin-autophagy or chromatophagy, where autolysosome fused with nuclear membrane and “extracts” broken chromatin out of nucleus[39], eventually leading to caspase-independent cell death. This was observed in prostate cancer cells[39], breast cancer cells[26]; hepatocellular carcinoma cells[57] and pancreatic cells[58].
In general, arginine deprivation initially induces autophagy to protect cells from starvation and at the same time, generates ROS (due to mitochondria impairment) and DNA damages which trigger apoptosis. During this early phase, an autophagy inhibitor such as chloroquine would increase cell death and enhances the drug efficacies[58]. For some cancer cells, however, autophagy persists and captures damaged broken DNA, leading to nuclear DNA leakage and cell death[28].
6.4. Necroptosis
As described above, arginine-deprivation induces autophagy which initially exerts a protective role and co-treatment with the autophagy inhibitor, chloroquine, and can facilitate the cell death[59][48][51][55]. In one study[48], it was shown such a treatment activates RIP kinase cascade, leading to necroptosis. Genetical knock-down of RIP1 or RIP3 or pharmaceutical treatment with necroptosis inhibitor, necrostatin, can protect against the co-treatment mediated cell death[51].
References
- Lindsey K. Boroughs; Ralph J. DeBerardinis; Metabolic pathways promoting cancer cell survival and growth. Nature 2015, 17, 351-359, 10.1038/ncb3124.
- Javier Garcia-Bermudez; Robert Williams; Rohiverth Guarecuco; Kıvanç Birsoy; Targeting extracellular nutrient dependencies of cancer cells. Molecular Metabolism 2019, 33, 67-82, 10.1016/j.molmet.2019.11.011.
- Songyun Zou; Xiangmei Wang; Po Liu; Changneng Ke; Shi Xu; Arginine metabolism and deprivation in cancer therapy. Biomedicine & Pharmacotherapy 2019, 118, 109210, 10.1016/j.biopha.2019.109210.
- Christin Riess; Fatemeh Shokraie; Carl Friedrich Classen; Bernd Kreikemeyer; Tomas Fiedler; Christian Junghanss; Claudia Maletzki; Arginine-Depleting Enzymes – An Increasingly Recognized Treatment Strategy for Therapy-Refractory Malignancies. Cellular Physiology and Biochemistry 2018, 51, 854-870, 10.1159/000495382.
- Leonard C. Rogers; Brian A. Van Tine; Innate and adaptive resistance mechanisms to arginine deprivation therapies in sarcoma and other cancers. Cancer Drug Resistance 2019, 2, 516-526, 10.20517/cdr.2019.49.
- Barbara Delage; Dean A. Fennell; Linda Nicholson; Iain McNeish; Nick Lemoine; Tim Crook; Peter W. Szlosarek; Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. International Journal of Cancer 2010, 126, 2762-2772, 10.1002/ijc.25202.
- Livingstone Fultang; Ashley Vardon; Carmela De Santo; Francis Mussai; Molecular basis and current strategies of therapeutic arginine depletion for cancer. International Journal of Cancer 2016, 139, 501-509, 10.1002/ijc.30051.
- Sofna Banjarnahor; Roman N. Rodionov; Jörg König; Renke Maas; Transport of L-Arginine Related Cardiovascular Risk Markers. Journal of Clinical Medicine 2020, 9, 3975, 10.3390/jcm9123975.
- Weimin Wang; Weiping Zou; Amino Acids and Their Transporters in T Cell Immunity and Cancer Therapy. Molecular Cell 2020, 80, 384-395, 10.1016/j.molcel.2020.09.006.
- Shuyu Wang; Zhi-Yang Tsun; Rachel L. Wolfson; Kuang Shen; Gregory A. Wyant; Molly E. Plovanich; Elizabeth D. Yuan; Tony D. Jones; Lynne Chantranupong; William Comb; et al.Tim WangLiron Bar-PeledRoberto ZoncuChristoph StraubChoah KimJiwon ParkBernardo L. SabatiniDavid M. Sabatini Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188-194, 10.1126/science.1257132.
- Tsung-Chin Lin; Yun-Ru Chen; Elizabeth Kensicki; Angela Ying-Jian Li; Mei Kong; Yang Li; Robert P. Mohney; Han-Ming Shen; Bangyan Stiles; Noboru Mizushima; et al.Liang-In LinDavid K. Ann Autophagy. Autophagy 2012, 8, 1477-1493, 10.4161/auto.21228.
- Rigel J. Kishton; Madhusudhanan Sukumar; Nicholas P. Restifo; Arginine Arms T Cells to Thrive and Survive. Cell Metabolism 2016, 24, 647-648, 10.1016/j.cmet.2016.10.019.
- Paulo C. Rodriguez; David G. Quiceno; Augusto C. Ochoa; l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2006, 109, 1568-1573, 10.1182/blood-2006-06-031856.
- Hidetoshi Ban; Kaori Shigemitsu; Tomoki Yamatsuji; Minoru Haisa; Tohru Nakajo; Munenori Takaoka; Tetsuji Nobuhisa; Mehmet Gunduz; Noriaki Tanaka; Yoshio Naomoto; et al. Arginine and Leucine regulate p70 S6 kinase and 4E-BP1 in intestinal epithelial cells. International Journal of Molecular Medicine 2004, 13, 537-543, 10.3892/ijmm.13.4.537.
- Robert A. Saxton; David M. Sabatini; mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960-976, 10.1016/j.cell.2017.02.004.
- Jenna L. Jewell; Kun-Liang Guan; Nutrient signaling to mTOR and cell growth. Trends in Biochemical Sciences 2013, 38, 233-242, 10.1016/j.tibs.2013.01.004.
- Jenna L. Jewell; Ryan Russell; Kun-Liang Guan; Amino acid signalling upstream of mTOR. Nature Reviews Molecular Cell Biology 2013, 14, 133-139, 10.1038/nrm3522.
- David Shahbazian; Philippe Roux; Virginie Mieulet; Michael S Cohen; Brian Raught; Jack Taunton; John W B Hershey; John Blenis; Mario Pende; Nahum Sonenberg; et al. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. The EMBO Journal 2006, 25, 2781-2791, 10.1038/sj.emboj.7601166.
- Bernadette Carroll; Dorothea Maetzel; Oliver Maddocks; Elsje Otten; Matthew Ratcliff; Graham R. Smith; Elaine A. Dunlop; Joao Passos; Owen Davies; Rudolf Jaenisch; et al.Andrew R. TeeSovan SarkarViktor I. Korolchuk Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife 2016, 5, e11058, 10.7554/elife.11058.
- Asier González; Michael N Hall; Nutrient sensing and TOR signaling in yeast and mammals. The EMBO Journal 2017, 36, 397-408, 10.15252/embj.201696010.
- Xiangfeng Kong; Bie Tan; Yulong Yin; Haijun Gao; Xilong Li; Laurie A. Jaeger; Fuller W. Bazer; Guoyao Wu; l-Arginine stimulates the mTOR signaling pathway and protein synthesis in porcine trophectoderm cells. The Journal of Nutritional Biochemistry 2012, 23, 1178-1183, 10.1016/j.jnutbio.2011.06.012.
- Lynne Chantranupong; Sonia M. Scaria; Robert A. Saxton; Melanie P. Gygi; Kuang Shen; Gregory A. Wyant; Tim Wang; Jeffrey Harper; Steven P. Gygi; David M. Sabatini; et al. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153-164, 10.1016/j.cell.2016.02.035.
- Chia-Lin Chen; Sheng-Chieh Hsu; Tan-Ya Chung; Cheng-Ying Chu; Hung-Jung Wang; Pei-Wen Hsiao; Shauh-Der Yeh; David K. Ann; Yun Yen; Hsing-Jien Kung; et al. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nature Communications 2021, 12, 1-14, 10.1038/s41467-021-22652-9.
- Thomas Porstmann; Claudio R. Santos; Beatrice Griffiths; Megan Cully; Mary Wu; Sally Leevers; John R. Griffiths; Yuen-Li Chung; Almut Schulze; SREBP Activity Is Regulated by mTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metabolism 2008, 8, 224-236, 10.1016/j.cmet.2008.07.007.
- Lei Shi; Xia Chen; Aiping Zang; Tiantian Li; Yanxiang Hu; Shixin Ma; Mengdie Lü; Huiyong Yin; Haikun Wang; XiaoMing Zhang; et al.Bei ZhangQibin LengJinbo YangHui Xiao TSC1/mTOR-controlled metabolic–epigenetic cross talk underpins DC control of CD8+ T-cell homeostasis. PLOS Biology 2019, 17, e3000420, 10.1371/journal.pbio.3000420.
- Fuming Qiu; Yun-Ru Chen; Xiyong Liu; Cheng-Ying Chu; Li-Jiuan Shen; Jinghong Xu; Shikha Gaur; Henry Jay Forman; Hang Zhang; Shu Zheng; et al.Yun YenJian HuangHsing-Jien KungDavid K. Ann Arginine Starvation Impairs Mitochondrial Respiratory Function in ASS1-Deficient Breast Cancer Cells. Science Signaling 2014, 7, ra31-ra31, 10.1126/scisignal.2004761.
- Chun-Ting Cheng; Yue Qi; Yi-Chang Wang; Kevin Chi; Yiyin Chung; Ching Ouyang; Yun-Ru Chen; Myung Eun Oh; Xiangpeng Sheng; Yulong Tang; et al.Yun-Ru LiuH. Helen LinChing-Ying KuoDustin SchonesChristina M. VidalJenny C.-Y. ChuHung-Jung WangYu-Han ChenKyle M. MillerPeiguo ChuYun YenLei JiangHsing-Jien KungDavid K. Ann Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Communications Biology 2018, 1, 1-15, 10.1038/s42003-018-0178-4.
- Sheng-Chieh Hsu; Chia-Lin Chen; Mei-Ling Cheng; Cheng-Ying Chu; Chun A. Changou; Yen-Ling Yu; Shauh-Der Yeh; Tse-Chun Kuo; Cheng-Chin Kuo; Chih-Pin Chuu; et al.Chien-Feng LiLu-Hai WangHong-Wu ChenYun YenDavid K. AnnHung-Jung WangHsing-Jien Kung Arginine starvation elicits chromatin leakage and cGAS-STING activation via epigenetic silencing of metabolic and DNA-repair genes. Theranostics 2021, 11, 7527-7545, 10.7150/thno.54695.
- Chun-Ting Cheng; Ching-Ying Kuo; Ching Ouyang; Chien-Feng Li; Yiying Chung; David C. Chan; Hsing-Jien Kung; David K. Ann; Metabolic Stress-Induced Phosphorylation of KAP1 Ser473 Blocks Mitochondrial Fusion in Breast Cancer Cells. Cancer Research 2016, 76, 5006-5018, 10.1158/0008-5472.can-15-2921.
- Issam Ben-Sahra; Jessica Howell; John M. Asara; Brendan D. Manning; Stimulation of de Novo Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Science 2013, 339, 1323-1328, 10.1126/science.1228792.
- Aaron M. Robitaille; Stefan Christen; Mitsugu Shimobayashi; Marion Cornu; Luca L. Fava; Suzette Moes; Cristina Prescianotto-Baschong; Uwe Sauer; Paul Jenoe; Michael N. Hall; et al. Quantitative Phosphoproteomics Reveal mTORC1 Activates de Novo Pyrimidine Synthesis. Science 2013, 339, 1320-1323, 10.1126/science.1228771.
- Shiran Rabinovich; Lital Adler; Keren Yizhak; Alona Sarver; Alon Silberman; Shani Agron; Noa Stettner; Qin Sun; Alexander Brandis; Daniel Helbling; et al.Stanley KormanShalev ItzkovitzDavid DimmockIgor UlitskySandesh C. S. NagamaniEytan RuppinAyelet Erez Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379-383, 10.1038/nature15529.
- Stephanie S. Kim; Shili Xu; Jing Cui; Soumya Poddar; Thuc M. Le; Hovhannes Hayrapetyan; Luyi Li; Nanping Wu; Alexandra M. Moore; Lei Zhou; et al.Alice C. YuAmanda M. DannIrmina A. ElliottEvan R. AbtWoosuk KimDavid W. DawsonCaius G. RaduTimothy R. Donahue Histone deacetylase inhibition is synthetically lethal with arginine deprivation in pancreatic cancers with low argininosuccinate synthetase 1 expression. Theranostics 2020, 10, 829-840, 10.7150/thno.40195.
- Anthony E. Pegg; Functions of Polyamines in Mammals. Journal of Biological Chemistry 2016, 291, 14904-14912, 10.1074/jbc.r116.731661.
- Chih-Ying Lee; Guan-Chin Su; Wen-Chi Huang; Min-Yu Ko; Hsin-Yi Yeh; Geen-Dong Chang; Sung-Jan Lin; Peter Chi; Promotion of homology-directed DNA repair by polyamines. Nature Communications 2019, 10, 65, 10.1038/s41467-018-08011-1.
- Ronald D. Snyder; Phillip J. Lachmann; Hyperthermia, Polyamine Depletion, and Inhibition of X-Ray-Induced DNA Strand Break Repair. Radiation Research 1989, 120, 121, 10.2307/3577639.
- Ronald D. Snyder; Prasad S. Sunkara; EFFECT OF POLYAMINE DEPLETION ON DNA DAMAGE and REPAIR FOLLOWING UV IRRADIATION OF HeLa CELLS. Photochemistry and Photobiology 1990, 52, 525-532, 10.1111/j.1751-1097.1990.tb01795.x.
- Matthew Locke; Essam Ghazaly; Maria Perpétua Freitas; Mikaella Mitsinga; Laura Lattanzio; Cristiana Lo Nigro; Ai Nagano; Jun Wang; Claude Chelala; Peter Szlosarek; et al.Sarah A. Martin Inhibition of the Polyamine Synthesis Pathway Is Synthetically Lethal with Loss of Argininosuccinate Synthase 1. Cell Reports 2016, 16, 1604-1613, 10.1016/j.celrep.2016.06.097.
- Chun A. Changou; Yun-Ru Chen; Li Xing; Yun Yen; Frank Y. S. Chuang; R. Holland Cheng; Richard J. Bold; David K. Ann; Hsing-Jien Kung; Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proceedings of the National Academy of Sciences 2014, 111, 14147-14152, 10.1073/pnas.1404171111.
- Ralph Scully; Arvind Panday; Rajula Elango; Nicholas A. Willis; DNA double-strand break repair-pathway choice in somatic mammalian cells. Nature Reviews Molecular Cell Biology 2019, 20, 698-714, 10.1038/s41580-019-0152-0.
- Vincenzo Bronte; Paola Zanovello; Regulation of immune responses by L-arginine metabolism. Nature Reviews Immunology 2005, 5, 641-654, 10.1038/nri1668.
- Sidney M. Morris; Sidney M.; Jr.; Arginine: Master and Commander in Innate Immune Responses. Science Signaling 2010, 3, pe27-pe27, 10.1126/scisignal.3135pe27.
- Paulo C. Rodriguez; Arnold H. Zea; Kirk S. Culotta; Jovanny Zabaleta; Juan B. Ochoa; Augusto C. Ochoa; Regulation of T Cell Receptor CD3ζ Chain Expression byl-Arginine. Journal of Biological Chemistry 2002, 277, 21123-21129, 10.1074/jbc.m110675200.
- Meera Rath; Ingrid Mã¼Ller; Pascale Kropf; Ellen Closs; Markus Munder; Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Frontiers in Immunology 2014, 5, 532-532, 10.3389/fimmu.2014.00532.
- Laura Poillet-Perez; Xiaoqi Xie; Le Zhan; Yang Yang; Daniel W. Sharp; Zhixian Sherrie Hu; Xiaoyang Su; Anurag Maganti; Cherry Jiang; Wenyun Lu; et al.Haiyan ZhengMarcus W. BosenbergJanice M. MehnertJessie Yanxiang GuoEdmund LattimeJoshua D. RabinowitzEileen White Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569-573, 10.1038/s41586-018-0697-7.
- Elena Brin; Katherine Wu; Hsin-Tze Lu; Yudou He; Zhaoming Dai; Wei He; PEGylated arginine deiminase can modulate tumor immune microenvironment by affecting immune checkpoint expression, decreasing regulatory T cell accumulation and inducing tumor T cell infiltration. Oncotarget 2017, 8, 58948-58963, 10.18632/oncotarget.19564.
- Peter W. Szlosarek; Astero Klampatsa; Arben Pallaska; Michael Sheaff; Paul Smith; Tim Crook; Matthew J. Grimshaw; Jeremy P. Steele; Robin M. Rudd; Frances Balkwill; et al.Dean A. Fennell In vivo Loss of Expression of Argininosuccinate Synthetase in Malignant Pleural Mesothelioma Is a Biomarker for Susceptibility to Arginine Depletion. Clinical Cancer Research 2006, 12, 7126-7131, 10.1158/1078-0432.ccr-06-1101.
- B Delage; P Luong; L Maharaj; C O’Riain; N Syed; T Crook; Eleftheria Hatzimichael; A Papoudou-Bai; T J Mitchell; S J Whittaker; et al.R CerioJ GribbenN LemoineJ BomalaskiC-F LiS JoelJ FitzGibbonL-T ChenP W Szlosarek Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death & Disease 2012, 3, e342-e342, 10.1038/cddis.2012.83.
- Tawnya L. Bowles; Randie Kim; Joseph Galante; Colin M. Parsons; Subbulakshmi Virudachalam; Hsing-Jien Kung; Richard J. Bold; Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. International Journal of Cancer 2008, 123, 1950-1955, 10.1002/ijc.23723.
- Jennifer X. Ji; Dawn R. Cochrane; Basile Tessier-Cloutier; Shary Yuting Chen; Germain Ho; Khyatiben V. Pathak; Isabel N. Alcazar; David Farnell; Samuel Leung; Angela Cheng; et al.Christine ChowShane ColborneGian Luca NegriFriedrich KommossAnthony KarnezisGregg B. MorinJessica N. McAlpineC. Blake GilksBernard E. WeissmanJeffrey M. TrentLynn HoangPatrick PirrotteYemin WangDavid G. Huntsman Arginine Depletion Therapy with ADI-PEG20 Limits Tumor Growth in Argininosuccinate Synthase–Deficient Ovarian Cancer, Including Small-Cell Carcinoma of the Ovary, Hypercalcemic Type. Clinical Cancer Research 2020, 26, 4402-4413, 10.1158/1078-0432.ccr-19-1905.
- Gregory R Bean; Jeff C Kremer; Bethany C Prudner; Aaron D Schenone; Juo-Chin Yao; Matthew B Schultze; David Chen; Munir Tanas; Douglas R Adkins; John Bomalaski; et al.Brian P RubinLoren S MichelBrian A Van Tine A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death & Disease 2016, 7, e2406-e2406, 10.1038/cddis.2016.232.
- Eun-Joo Noh; Sang-Wook Kang; Yong-Jae Shin; Sang-Hyun Choi; Chan-Gil Kim; In-Sun Park; Denys N. Wheatley; Bon-Hong Min; Arginine deiminase enhances dexamethasone-induced cytotoxicity in human T-lymphoblastic leukemia CCRF-CEM cells. International Journal of Cancer 2004, 112, 502-508, 10.1002/ijc.20435.
- Hui Jiang; Song Guo; Dan Xiao; Xuzhao Bian; Jie Wang; Ying Wang; Huiting Zhou; Jun Cai; Zhongliang Zheng; Arginine deiminase expressed in vivo, driven by human telomerase reverse transcriptase promoter, displays high hepatoma targeting and oncolytic efficiency. Oncotarget 2017, 8, 37694-37704, 10.18632/oncotarget.17032.
- Niramol Savaraj; Chunjing Wu; Marcus Tien Kuo; Min You; Medhi Wangpaichitr; Carlos Robles; Seth Spector; Lynn Feun; The Relationship of Arginine Deprivation, Argininosuccinate Synthetase and Cell Death in Melanoma. Drug Target Insights 2007, 2, 119-128, 10.1177/117739280700200016.
- N Syed; J Langer; K Janczar; P Singh; Cristiana Lo Nigro; L Lattanzio; H M Coley; E Hatzimichael; J Bomalaski; P Szlosarek; et al.M AwadK O'NeilF RoncaroliT Crook Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death & Disease 2013, 4, e458-e458, 10.1038/cddis.2012.197.
- M P Kelly; A A Jungbluth; B-W Wu; J Bomalaski; L J Old; G Ritter; Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. British Journal of Cancer 2011, 106, 324-332, 10.1038/bjc.2011.524.
- Qingyuan Feng; Xuzhao Bian; Xuan Liu; Ying Wang; Huiting Zhou; Xiaojing Ma; Chunju Quan; Yi Yao; Zhongliang Zheng; Intracellular expression of arginine deiminase activates the mitochondrial apoptosis pathway by inhibiting cytosolic ferritin and inducing chromatin autophagy. BMC Cancer 2020, 20, 1-13, 10.1186/s12885-020-07133-4.
- Nathalie Khalil; Ralph J. Abi-Habib; [HuArgI (co)-PEG5000]-induced arginine deprivation leads to autophagy dependent cell death in pancreatic cancer cells. Investigational New Drugs 2019, 38, 1236-1246, 10.1007/s10637-019-00883-4.
- Randie Kim; Jodi M. Coates; Tawnya L. Bowles; Gregory P. McNerney; Julie Sutcliffe; Jae U. Jung; Regina Gandour-Edwards; Frank Y.S. Chuang; Richard J. Bold; Hsing-Jien Kung; et al. Arginine Deiminase as a Novel Therapy for Prostate Cancer Induces Autophagy and Caspase-Independent Apoptosis. Cancer Research 2009, 69, 700-708, 10.1158/0008-5472.can-08-3157.