1. Clinical Features of Chronic Myeloid Leukemia
Chronic myeloid leukemia (CML) is a myeloproliferative disease with an incidence of 1–2 cases per 100,000 each year, accounting for 15% of all new cases of leukemia
[1]. The frequency is higher among adults, in whom the mean age of incidence is about 55 years, and indeed, rarely arises during childhood. It may affect both sexes, but is slightly more common in males, with a ratio of 2.2 men to 1.4 women per 100,000 affected
[1][2][1,2]. The most common clinical symptoms of CML include fatigue, anemia, splenomegaly, abdominal pain, and recurrent infections. However, a large proportion of asymptomatic patients are diagnosed after an unrelated medical examination
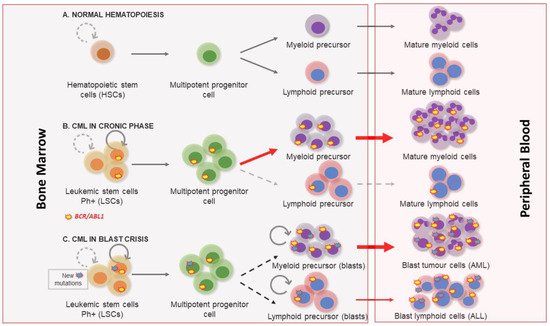
[1]. Three clinical phases of its pathological evolution are recognized. At first, CML disease is characterized by a myeloid hyperplasia in an indolent chronic phase (CP). At this point, leukemic stem cells (LSCs) respond to growth factors, but myeloproliferative differentiation pathways acquire an advantage because they are the main cause of the massive myeloid expansion characteristic of CML
[3]. In this initial phase, myeloid progenitors and mature cells accumulate in the blood and extramedullary tissues. Without effective therapy, CML progresses through a period of increasing instability known as the acceleration phase (AP), ending in an acute leukemic-like disease known as the blast crisis phase (BP). The definitions of AP and BP are largely dependent on the proportion of blasts in the blood and bone marrow. AP and BP are characterized by a maturation arrest in the myeloid or lymphoid lineage, and newly accumulated genetic and epigenetic aberrations occur in LSCs
[4]. The final BP stage can result in a lymphoblastic (25%), myeloblastic (50%), or biphenotypic/undifferentiated acute leukemic phenotype (25%), which indicates a stem origin for CML disease
[5], as shown in . Finally, bone marrow failure due to a lack of cell differentiation, and a massive infiltration by immature blasts, causes patient mortality from infection, thrombosis, or anemia
[6].
Figure 1. Chronic myeloid leukemia clinical phases. (A) Normal hematopoiesis characterized by the existence of hematopoietic stem cells with a controlled self-renewal and multipotency ability, resulting in balanced hematopoiesis between myeloid and lymphoid lineages. (B) In the chronic phase the myeloproliferative differentiation pathway acquires an advantage, and a massive myeloid expansion is produced. (C) Blast crisis is characterized by a maturation arrest in the myeloid or lymphoid lineage. Newly accumulated genetic and epigenetic aberrations appear in leukemic stem cells (LSCs) and blast cells go out from bone marrow to peripheral blood.
Diagnosis is based on detecting the hallmark of CML, the presence of the chromosome 22 abnormality known as Philadelphia (Ph), named after the US city in which it was first observed. It is the result of the reciprocal translocation between chromosomes 9 and 22-t(9;22)
[7]. Conventional cytogenetics, fluorescence in situ hybridization (FISH), and reverse transcription PCR (RT-PCR) are the techniques commonly used to confirm a diagnosis of CML and to evaluate the response to therapy.
Before successful treatments became available, the median survival of CML patients after diagnosis was approximately 3–5 years
[8][9][8,9]. The therapeutic landscape of CML changed profoundly with the introduction of tyrosine kinase inhibitor (TKI) drugs
[8][10][11][8,10,11], and most patients with CP-CML now have a normal life expectancy. However, treatment discontinuation is only an option for a small subset of patients
[12].
2. Molecular Biology of Chronic Myeloid Leukemia
Nowell and Hungerf, in 1960, first described the Ph chromosome, a small chromosome present in the bone marrow cells of CML patients
[7]. It was the first time that a chromosomal abnormality had been linked to a particular neoplasia
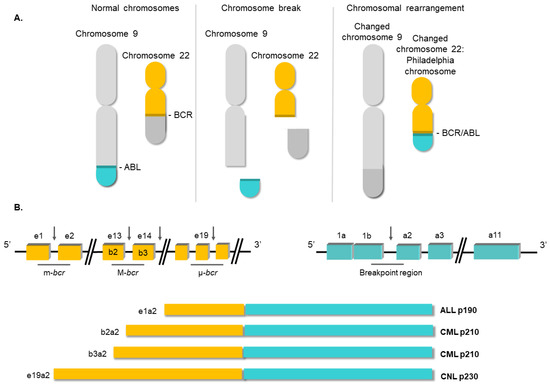
[13]. Subsequent investigations confirmed that the generation of the Ph chromosome was due to the t(9;22) (q34;q11) translocation. The next breakthrough in our understanding of CML occurred in the 1980s, when it was demonstrated that this rearrangement gave rise to a fusion gene
[14]. In this translocation, the analogue of the v-
ABL protooncogene from chromosome 9 is moved to the breakpoint cluster region of the
BCR gene on ch22. The location of the breakpoints between the two loci is variable
[15]. Commonly, the breakpoint at the
ABL locus occurs in a DNA region spanning more than 200 kb housing exon 2. At the
BCR locus, the breakpoints occur in the major breakpoint cluster region (M-
bcr), which spans a 3 kb region that includes exons 13 and 14 of
BCR. All the rearrangements involving both breakpoint regions give rise to a 210 kDa protein, the most common chimeric transcript in CML
[16]. However, in a minority of CML cases, the
BCR breakpoint is located near exon 2, termed the minor breakpoint cluster region (m-
bcr). In these cases, the resulting mRNA gives rise to a 190 kDa protein
[15]. Finally, another infrequent breakpoint cluster region (μ-
bcr) exists, downstream of
BCR exon 19, which generates a 230 kDa protein when it is translocated to the
ABL1 locus
[17], as shown in .
Figure 2. Structure of the BCR/ABL1 oncogene. (A) Schematic representation of the t(9;22) (q34;q11) translocation triggering the Philadelphia chromosome. (B) Breakpoint locations between BCR and ABL1 genes. Different fusion protein combinations yield different outcomes.
Since the
BCR/ABL1 fusion was described, the efforts of the scientific community have focused on elucidating its molecular roles in CML pathology. Several studies have shown the aberrant and constitutive tyrosine kinase activity of the
BCR/ABL1 oncoprotein, highlighting this activity as being responsible for the transformation of the hematopoietic stem cell
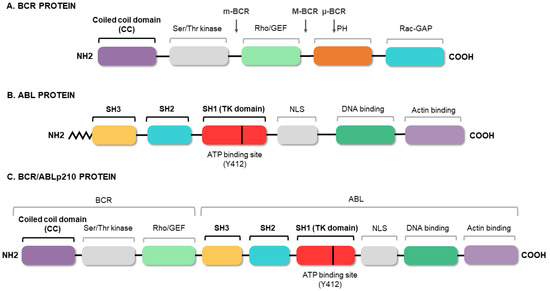
[18][19][20][21][18,19,20,21]. The fusion of the two genes constitutively activates the tyrosine kinase domain of
ABL1, which contains three SRC homology domains (SH1–SH3). The SH1 domain enables the tyrosine kinase function, whereas the SH2 and SH3 domains mediate interactions with other proteins
[22]. The SH3 domain is critical to the regulation of
ABL1 kinase activity, thereby presenting a target for clinical therapy. It is known that the fusion between the 5′ end of
BCR and the SH3 domain of
ABL1 abrogates the physiological suppression of the kinase
[23]. Meanwhile,
BCR has an important coiled-coil (CC) domain that will allow
BCR/ABL1 dimerization and subsequent trans-autophosphorylation, thus increasing the molecular signal
[24], as shown in . The phosphorylation of the Y-177 tyrosine residue domain SH2 of
ABL allows the high-affinity binding of the growth factor receptor-bound protein 2 (GRB2) as well as the scaffolding protein Gab2, activating the Ras pathway
[25]. This aberrant kinase signaling activates many target proteins, such as the PI3K, AKT, JNK, and SRC family kinases, as well as transcription factors such as STATs, nuclear factor-κB, and MYC
[26][27][28][26,27,28].
Figure 3. BCR/ABL protein domains. Protein regions located in the BCR (A) and ABL (B) proteins, and those maintained in the fusion (C). The figure highlights the coiled-coil (CC) domain of BCR, which allows the dimerization of the oncoprotein, and the three SRC domains of ABL1, including the tyrosine kinase domain (SH1) and the regulatory domains (SH2 and SH3).
The constitutively active signaling causes cell reprogramming and expansion of the LSC clone. As a result,
BCR/ABL1-positive hematopoietic stem cells exhibit uncontrolled proliferation
[29], lack of response to apoptotic signals
[30], alterations in cell adhesion
[31], impaired differentiation
[32], and independence of growth factors
[33]. As a consequence, a myeloid differentiation bias is commonly observed in the chronic phase of CML.
3. Conventional Therapies for Chronic Myeloid Leukemia
The history of CML treatment can be considered one of the great milestones of modern cancer medicine. From its discovery until the 1980s, the standard treatment for CML consisted of conventional chemotherapy. Arsenic was the first treatment to be administered in the 19th century, but was superseded by alkylating drugs such as busulfan and hydroxyurea in the 1960s
[34][35][34,35]. Unfortunately, they did not delay the onset of disease progression and facilitated only a modest improvement in survival. The introduction of interferon-α in the 1970s induced complete cytogenetic remission in 10–15% of patients, and increased median survival to 6 years
[36]. However, interferon-α treatment has serious side-effects, and treatment had to be discontinued in most patients, causing them to relapse. In this context, allogenic stem cell transplantation (SCT) was the only therapeutic option that could provide increased long-term survival, and so it became the first-line treatment in the 1990s for patients in the chronic phase
[37][38][39][37,38,39]. Even today, this therapeutic option is the only one with the potential to definitively cure CML patients in this phase. The SCT procedure involves bone marrow ablation (by chemotherapy or radiotherapy) followed by the infusion of normal allogenic stem cells. However, it is only available to a small number of patients who have an HLA-matched donor, and is associated with a significant transplant-related mortality rate
[39]. Nowadays, SCT is used solely as a last-resort salvage option.
As mentioned above, CML is a type of cancer in which all the pathological features can be attributed to a single genetic event, in this case the
BCR/ABL1 fusion. Knowing that the tyrosine kinase activity of
BCR/ABL1 is essential for the malignant transformation of cells, the search for compounds that inhibit this activity became imperative. During the 1990s, various tyrosine kinase inhibitors (TKIs) were tested to evaluate their therapeutic potential in CML
[40][41][40,41]. The mechanism of action of these compounds is based on competition with adenosine triphosphate (ATP) or the protein substrate of the kinase, whereby
BCR/ABL1 activity is inhibited at the protein level. Finally, in the 2000s, the Novartis compound STI571 (later known as imatinib mesylate), which showed surprising results by selectively inhibiting
BCR/ABL1 at micromolar concentrations, was approved as therapy for CML
[42][43][42,43]. The arrival of TKIs marked a watershed in the treatment of CML and they remain the frontline therapy for CML. Thanks to TKIs, CP-CML patients, who, before 2001, had a survival rate of 20% at 8 years, now have a rate of 87%, and a life expectancy like those of healthy people of the same age
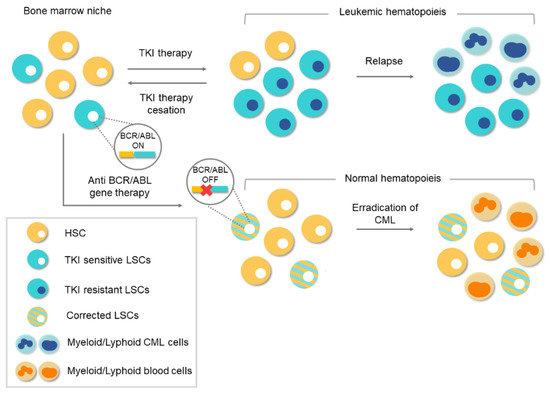
[10][11][17][10,11,17]. Despite the success achieved with TKI-based treatments, there are still obstacles to overcome. The main concern is that TKI drugs do not tackle the etiological cause of CML and the oncogenic event remains uncorrected/unedited. The existence of residual
BCR/
ABL-positive cells, which remain “oncogenic-quiescent”, has been demonstrated, indicating that TKIs do not completely eliminate the LSCs
[12]. TKIs efficiently silence the oncogenic activity of
BCR/
ABL while the drug is present, but the remaining LSCs can lead to relapse after TKI therapy ceases, as shown in .
Figure 4. Conventional therapies vs. gene therapy for chronic myeloid leukemia (CML). Tyrosine-kinase inhibitor (TKI)-based conventional therapies are effective at silencing BCR/ABL1 in leukemic stem cells (LSCs). Treatment cessation can lead to relapse because of the existence of residual BCR/ABL1-positive cells. The appearance of TKI-resistant LSCs during treatment can lead to a relapse of the disease. However, anti-BCR/ABL1 gene therapy would eliminate the oncogene at the genome level. Corrected LSCs would be able to repopulate the bone marrow niche and thereby enable normal hematopoiesis.
In this scenario, lifelong oral medication is necessary, and treatment discontinuation is only an option in those patients who were able to achieve and maintain strong molecular responses. Lifelong administration facilitates adverse effects in many patients and a significant percentage of them eventually become resistant to TKI treatment
[44]. The identification of various forms of resistance has led to the development of second- and third-generation TKIs that are effective against kinase-specific mutations in these patients
[45]. Taking this therapeutic scenario into account, it is still necessary to seek new and definitive alternative therapies. Currently, any coding sequence can be abolished by CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 nucleases
[46][47][48][46,47,48] or zinc finger nuclease
[49], which means there is an opportunity of a definitive cure available to TKI-resistant CML patients. Thus, CRISPR/Cas9 system could be a definitive therapeutic option.