Primary central nervous system lymphoma (PCNSL) is one of a few lymphomas that primarily arise in “immune sanctuary/immune-privileged” sites. It is a rare form of extranodal non-Hodgkin lymphoma that primarily arises in the brain, spinal cord, leptomeninges, and vitreoretinal compartment of the eye and shows no significant systemic involvement.
- primary central nervous system lymphoma (PCNSL)
- diffuse large B-cell lymphoma
1. Epidemiology and Clinical presentation
The terms ‘PCNSL’ and ‘primary central nervous system diffuse large B-cell lymphoma’ (PCNS DLBCL) have been used interchangeably, as 90–95% of PCNSL are DLBCL. Because of the latter, our discussion of epidemiologic data, and clinical presentation are most applicable to PCNS DLBCL. PCNSL comprises 4–7% of all brain tumors [2][1], 5% of all extranodal lymphomas [3][2], and less than 1% of all non-Hodgkin lymphomas. It is the third most common malignancy that primarily arises in the central nervous system after glioblastoma and diffuse astrocytoma. In the general population, PCNSL had an annual incidence of 0.43 per 100,000 during the period of 2009–2013 [3][2]. There is a slight male predominance (ratio of 1.25) and a higher incidence in the Caucasian population in comparison to African American population (ratio of 1.33) [3,4][2][3].
Two important risk factors of PCNSL are increasing age and human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS), with a 3600-fold increased incidence of PCNSL ever reported in AIDS patients [4][3]. Currently, approximately 19% of all patients with PCNSL in United States have HIV [5][4] (Figure 1). This estimate has been declining since the introduction and widespread use of highly active antiretroviral therapy (HAART) [6][5]. Whereas the median age of PCNSL at diagnosis is 66 years [3][2], the diagnosis is usually made at a younger age in HIV/AIDS patients (median of 40.7 years) [7][6]. Compared to other non-Hodgkin lymphomas, HIV/AIDS patients with PCNSL have the lowest CD4 count (median of 14 cells/µL) at diagnosis [7][6], reiterating that immunodeficiency is a significant risk factor for developing PCNSL.
Patients with PCNSL present with varying symptoms according to the central nervous system (CNS) compartment involved. Brain involvement can result in focal neurological deficits, neuropsychiatric symptoms, seizures, and increased intracranial pressure manifesting as headache, nausea, and vomiting [8,9][7][8]. Among the aforementioned symptoms, seizure is relatively less common because the cortex is less commonly involved by PCNSL [9][8]. Spinal involvement by PCNSL usually manifests as discrete intramedullary nodules, and the symptomatology (asymmetric sensory changes, weakness in extremities, and bowel/urinary bladder dysfunction) is similar to that of other intramedullary tumors [9][8]. Involvement of the peripheral nerve system that includes the peripheral nerves, nerve roots, plexus, and cranial nerves is referred as neurolymphomatosis [10][9]. It can present with painful peripheral neuropathy or radiculopathy, cranial neuropathy, painless polyneuropathy, and peripheral mononeuropathy or a mononeuropathy multiplex [10][9]. Ocular involvement by PCNSL, which is seen in 20–25% of cases [11][9], can result in decreased visual acuity, blurry vision, and/or floaters [9][8]. Contralateral tumors and parenchymal CNS involvement are seen in a majority of patients (80–90%) with intraocular tumors [12][10].
The preferred and most sensitive imaging modality for the evaluation of brain parenchyma in the context of PCNSL is the gadolinium-enhanced magnetic resonance imaging (MRI) scan. Contrast-enhanced computed tomography (CT) scans are an appropriate alternative in patients where MRI scans are contraindicated or when MRI is unavailable [13][11]. On T1-weighted magnetic resonance (MR) images, PCNSL lesions are hypointense, whereas they are isointense to hyperintense on T2-weighted MR images [9,12][8][10] (Figure 2). The amount of peritumoral edema is usually less extensive than in malignant gliomas and metastases [12][10]. In a retrospective review of 248 cases of primary intracerebral malignant lymphoma by Bataille et al., a majority (66%) of the patients presented with a single lesion, and 90% of the lesions were larger than 1 cm [8][7]. Eighty-nine percent (175/196) of the lesions analyzed showed supratentorial involvement, and anatomic locations included (in decreasing orders of frequency) the frontal lobe (20%), parietal lobe (18%), temporal lobe (15%), basal ganglia (13%), corpus callosum (11%), brainstem (7%), cerebellum (6%), insula (4%), occipital lobe (4%), and fornix (3%) [8][7]. A bilateral mirror pattern was seen in 5% of Bataille’s cohort [8][7].
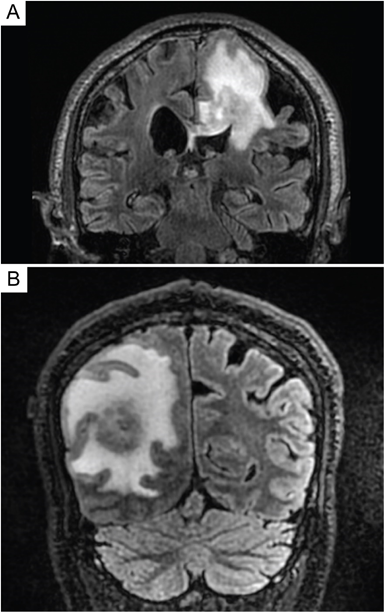
Figure 2. MRI images of a primary central nervous system lymphoma. T2-weighted-Fluid-Attenuated Inversion Recovery (T2-FLAIR) images of patients with primary central nervous system lymphoma that demonstrate T2 hyperintense lesions with increased signal within the surrounding parenchyma. Lesions may mimic primary glial tumors, and when involving the corpus callosum may overlap in appearance with glioblastoma (A). Patients may present with a solitary mass with significant peritumoral edema (B) or also present with multifocal intracranial disease.
2. Primary Central Nervous System Diffuse Large B-Cell Lymphoma (PCNS DLBCL)
Diffuse large B-cell lymphoma (DLBCL) accounts for 90–95% of all PCNSL [5]. PCNS DLBCL includes DLBCL arising within the brain, spinal cord, leptomeninges, and vitreo-retina [12]. DLBCL of the dura and ocular adnexa, intravascular large B-cell lymphomas, those of systemic disease (secondary lymphomas), and most immunodeficiency-associated lymphomas (e.g., in the posttransplant or iatrogenic-related setting) are excluded from this category [12]. The etiology of PCNS DLBCL in the immunocompetent population is still unknown. Viruses such as Epstein-Barr virus (EBV), human herpesvirus-6 (HHV-6) [24][13], human herpesvirus-8 (HHV-8) [25][14], and polyomaviruses [26,27][15][16] have been shown to play no causative role.
On gross/macroscopic examination, PCNS DLBCL shows a soft and pale, fish flesh appearance. It is characterized histomorphologically by a diffuse proliferation of medium-to-large-sized lymphoid cells with pleomorphic, round to oval, irregular, and vesicular nuclei with prominent nucleoli, morphologically consistent with centroblasts or immunoblasts (Figure 4A). Tumor cells usually exhibit perivascular arrangement (angiocentricity) by forming layers around blood vessels (Figure 4B). Similar to high-grade glial neoplasms, areas of geographic necrosis are commonly present in the center of the tumor. However, microvascular proliferation (frequently encountered in high-grade gliomas) is rare in primary CNS DLBCL and is often nonexistent [12]. A biopsy obtained from the periphery of the tumor could pose a diagnostic challenge, because the lymphomatous cells can infiltrate the brain parenchyma as single cells and can be easily obscured by the background astrogliosis and reactive inflammatory cells consisting of T-cells, B-cells, and foamy histiocytes [12]. Immunohistochemical stains for lymphoid markers should be performed if there is any suspicion for lymphoma. The presence of reactive perivascular T-cell infiltrate, defined as the presence of at least one small-to-medium-sized vessel surrounded by a rim of small-to-intermediate-sized T-cells, may correlate with favorable outcomes and should be described in the report when observed [28][17].
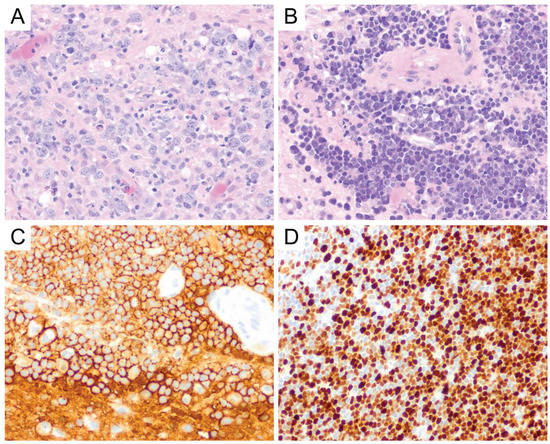
Figure 4. Primary CNS DLBCL. Lymphoma cells are round to oval and contain irregular, vesicular nuclei with prominent nucleoli, morphologically consistent with centroblasts or immunoblasts (A, original magnification ×400). Primary CNS DLBCL usually shows perivascular arrangement (angiocentricity) with tumor cells forming layers around the blood vessels (B, original magnification ×400). They express CD20 (C, original magnification ×400) and usually show a high Ki-67 proliferation index (D, original magnification ×400).
PCNS DLBCL is a mature B-cell neoplasm, and the tumor cells express B-cell markers, particularly CD19, CD20 (Figure 4C), CD22, CD79a, and PAX5 [12]. A majority (67–96%) [18][19][20][21] [29,30,31,32] of cases are of an activated B-cell (ABC)/non-germinal center B-cell (non-GCB) subtype where the tumor cells are either (i) CD10-negative, Bcl-6-negative or (ii) CD10-negative, Bcl-6-positive, and MUM1-positive by immunohistochemistry using the Hans algorithm [33][22]. The remainder of the cases are of a germinal center B-cell (GCB) subtype, which is defined by CD10 expression in at least 30% of tumor cells or Bcl-6 expression in the absence of MUM1 expression [33][22]. Bcl-6 and MUM1 are expressed in the majority of PCNS DLBCL, but CD10 expression is only seen in a small portion of cases (<10%) [1,12][1][12]. Therefore, CD10 expression should always prompt the investigation of a potential systemic DLBCL that has disseminated into the central nervous system. In a study by Montesinos-Rongen et al., reverse transcriptase-polymerase chain reaction (RT-PCR) for transcripts of immunoglobulin constant region gene segments, performed on 11 PCNS DLBCL samples, showed exclusive transcription of IgM and IgD mRNA in the absence of IgG, IgA, or IgE transcription [34][23]. Neoplastic Bcells can show either kappa or lambda light chain restriction, but plasma cell markers (i.e., CD138 and CD38) are usually negative [12]. The Ki-67 proliferation index in PCNS DLBCL is usually very high, up to 90% [24] [35] (Figure 4D), and it was suggested that the high proliferation index might correlate well with the frequently observed c-Myc expression in PCNS DLBCL. This hypothesis was supported by the observation that, in Brunn’s series, two of the three cases of primary CNS DLBCL with a relatively low Ki-67 proliferation index (i.e., <50%) also showed low c-Myc expression [35][24]. EBV expression in PCNS DLBCL is not common and may reflect an underlying immunodeficiency [12].
The worse prognosis of PCNS DLBCL in comparison to systemic DLBCL was first thought to be due to the significant portion of cases with an ABC subtype [30][19], which is generally associated with a poorer prognosis in comparison to the GCB subtype. However, some studies reported no significant difference in the overall survival and progression-free survival between the ABC and GCB subtypes of PCNS DLBCL [29,36][18][25]. Interphase cytogenetic analysis showed that IGH and BCL6 rearrangements were seen in, respectively, 13% and 17–26% of PCNS DLBCL [35,37,38][24][26][27]. Whereas systemic DLBCL with co-expression of c-Myc and Bcl-2 (double-expressor) and/or co-rearrangement of MYC and BCL2 and/or BCL6 (double or triple hit lymphoma) has been associated with a worse prognosis, the findings were inconclusive for PCNS DLBCL. The expression of c-Myc and Bcl-2 proteins has been described in a significant portion of PCNS DLBCL, respectively, up to 70–90% [24][25] [35,36] and 59–73% [28][29][30] [39,40,41] of cases. Interestingly, MYC gene rearrangements are significantly lower in frequency (3–8%) [35[24][26],37], and thus far, BCL2 gene rearrangements have not been reported [35,42] [24][31] in PCNS DLBCL. This suggests that increased c-Myc and Bcl-2 protein expressions might be attributable to other genetic aberrations, such as MYC and BCL2 gene mutations or altered regulation of expression (e.g., through post-transcriptional and post-translational modifications) independent of MYC or BCL2 gene rearrangements. Although some studies showed that positive c-Myc protein expression (defined as positive immunohistochemical staining in at least 40% of tumor cells) was associated with worse overall and progression-free survival [39[28][29][32],40,43], other studies found no significant difference in prognosis [29,36][18][25]. It is important to note that these studies used the same cutoff to define positive c-Myc protein expression. Tapia et al. used a threshold greater than 30% to define c-Myc positivity and reported the association of c-Myc expression with lower overall survival [41][30]. It is still unclear why these results have varied across the studies and it does not seem to be related to the c-Myc antibodies used. It needs to be noted that Kim’s group used c-Myc antibodies from Cell Marque and they reported a significantly lower c-Myc-positive cases (18.1%) in their cohort [40][29]. Similarly, inconclusive findings were observed for Bcl-2 protein expression. Shi et al. [39][28], Kim at al. [40][29], and Makino et al. [18] [29] reported a less favorable overall survival associated with high Bcl-2 protein expression, while Tapia et al. [41][30] and Liu et al. [33] [44] reported no significant difference in prognosis. It is unclear whether the inconclusive findings across the Bcl-2 studies were caused by different cutoffs used to define positivity for Bcl-2, which ranged from 50–70% [29,39,40,44][18][18][29][33]. However, Tapia et al. evaluated several cutoffs to define Bcl-2 positivity, 30%, 50%, and 70%, and found no significant association between Bcl-2 expression and prognosis [41][30]. Similarly, several studies reported a more adverse clinical outcome associated with co-expression of c-Myc and Bcl-2 [40,45][29][34] and other studies failed to demonstrate such an association [29,36][18][25]. Given these inconclusive data, the decision to perform immunohistochemistry for cell-of-origin status (i.e., GCB vs. ABC), double expressor status, and/or fluorescence in situ hybridization for MYC, BCL2, and BCL6 rearrangements may be highly variable in various practices and will at least be partly based upon the preference of the treating oncologists.
The advent of genetic analysis has expanded the knowledge in the histogenetic origin of PCNS DLBCL. For example, the presence of mutations in the 5’-noncoding region of the BCL6 gene (a marker of B-cell transition through the germinal center) in a significant subset of PCNS lymphomas suggests that this lymphoma might be derived from germinal center Bcells. Montesinos-Rongen et al. further showed that all PCNS DLBCLs in their series carried monoclonally rearranged V region genes and had introduced somatic mutations into the rearranged IG genes [46][35][36][37]. These findings suggest that PCNS DLBCLs are derived from mature B-cells with prior antigenic exposure and have undergone T-cell-dependent affinity maturation in the microenvironment of the germinal center of secondary lymphatic organs [46][35].
Despite their histomorphologic and immunophenotypic resemblance to systemic DLBCL, PCNS DLBCL might represent a distinct subtype that is characterized by their unique clinical and molecular features related to the CNS milieu. The immunologically privileged microenvironment of the CNS has been one of the subjects in the earlier studies of pathogenesis of PCNSL, and it was postulated that an interplay between chemokines and chemokine receptors or cytokines within the CNS microenvironment might play a key role in the development of PCNSL [12,47][12][36]. Interleukin (IL)-4, a B-cell growth and survival factor, has been found to be expressed by tumor cells and tumor vasculature in CNS lymphomas [48][37]. Several IL-4-induced gene products, including X-box binding protein 1 (XBP-1), have been found to be highly expressed in PCNSL [48][37]. XBP-1 regulates unfolded protein response (UPR) and both UPR and XBP-1 are essential for tumor growth under hypoxic stress and glucose deprivation [48][37]. In addition, the activated form of signal transducer and activator of transcription 6 (STAT6), a mediator of IL-4 signaling, was expressed by tumor cells and tumor endothelia in PCNSL, and a high expression of activated STAT6 was associated with shorter survival in patients with PCNSL who were treated with high-dose intravenous methotrexate therapy [48][37].
Aberrant and constitutive activation of nuclear factor (NF)-ĸB signaling pathway is a hallmark of PCNSL (Figure 5), which can be mediated through the gain of 18q21.33-q23, activating mutation of CARD11 (caspase recruitment domain family member 11) and stimulation of the B-cell receptor (BCR), tumor necrosis factor (TNF), or toll-like receptor (TLR) pathway [49][38]. The gain of 18q21.33-q23 has been described in 43% of PCNSL and is one of the most common chromosomal abnormalities seen in PCNSL [50][39]. Other commonly seen chromosomal abnormalities include the loss of 6q21 (52%) and 6p21.32 (37%) and gain of 19q13.43 (47%), 1q, 7, and 12 [50][39]. The CARD11 gene encodes for the CARD11 protein, a mediator of the BCR pathway [49][38]. The TLR pathway deregulation in PCNSL is commonly secondary to mutations in the myeloid differentiation primary response gene 88 (MYD88), which is highly recurrent in PCNSL and has been described in at least 50% of PCNSL [12,42,49][10][31][38]. This is at a higher rate than described for systemic DLBCL (10–20%) [42][31]. MYD88 is an adaptor protein that mediates TLR and IL-1 receptor signaling [51][40]. It associates with interleukin 1 receptor-associated kinase 1 (IRAK1) and IRAK4 to promote tumor survival [51][40]. In 71% of PCNSL with MYD88 mutations, a leucine to proline exchange at position 265 (L265P) has been found [49][38]. MYD88 L265P is an activating somatic mutation that promotes survival by spontaneously assembling a protein complex containing IRAK1 and IRAK4, ultimately leading to NF-ĸB signaling pathway activation, Janus kinase (JAK) activation of STAT3, and the secretion of IL-6, IL-10, and interferon-β [49,51][38][40]. IL-10 may play a pivotal role in suppressing immune reactions toward tumor cells, hence promoting their survival. Using whole-exome sequencing of 41 PCNS DLBCL cases, Fukumura et al. identified other frequently mutated genes in the NF-ĸB signaling pathway, including PIM1, BTG2, CD44, XBP1, CD79B, and NFKB1E [42][31]. Their study also showed that mutations in PIM1 (100%) and BTG2 (92.7%) were more frequently seen in PCNS DLBCL in comparison to systemic DLBCL [42][31]. In contrast, RHOH and BCL6, which are frequent targets of aberrant somatic hypermutation in systemic DLBCL, were not affected in PCNS DLBCL [42][31]. These findings lend further support for the notion that PCNS DLBCL represents a distinct entity from systemic DLBCL. In the same study, Fukumura et al. also examined prognostic values and potential therapeutic implications of their results. They demonstrated that alterations in HLA-C were associated with a shorter progression-free survival (PFS) and that activation of the genes at the 7q35 locus might contribute to PCNS DLBCL relapse [42][31]. In addition, chromosome copy number alterations involving TP53 were not associated with a shorter progression-free survival [42][31]. Furthermore, 6 of the 41 cases in the Fukumura’s series carried GRB2 mutations, a gene that codes for an adapter protein that binds to tyrosine kinases and other docking proteins through its Src homology 2 (SH2) domain and transduces growth signals through the RAS-MAPK pathway [42][31]. When GRB2(V140G)-expressing 3T3 cells were treated in vitro with MAP2K1/2 inhibitors (trametinib and selumetinib), their malignant transformation was attenuated [42][31], a promising result that merits further investigations.
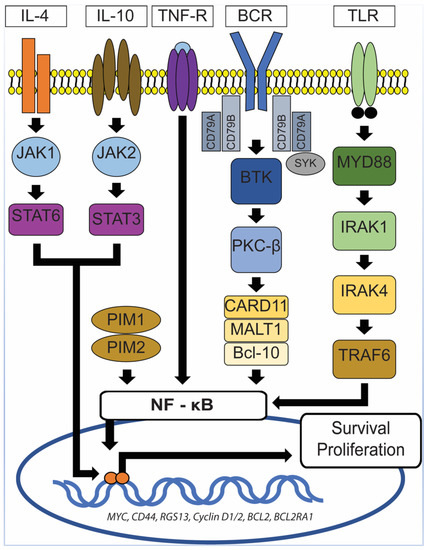
Figure 5. NF-ĸB signaling pathway. In PCNSL, activation of NF-ĸB signaling pathway can be mediated through the gain of 18q21.33-q23, activating the mutation of CARD11 and stimulation of the B-cell receptor (BCR), tumor necrosis factor (TNF), or toll-like receptor (TLR) pathway. MYD88 mutations, which are frequently seen in PCNSL, lead to spontaneous assembly of a protein complex containing IRAK1 and IRAK4, ultimately resulting in NF-ĸB signaling pathway activation, Janus kinase (JAK) activation of STAT3 pathway, and the secretion of IL-6, IL-10, and interferon-β. TNF-R: TNF receptor, MYD88: Myeloid differentiation primary response gene 88, CARD11: Caspase recruitment domain family member 11, IRAK1/4: Interleukin 1 receptor-associated kinase 1/4, TRAF6: Tumor necrosis factor receptor (TNFR)-associated factor 6, BCR: B-cell receptor, SyK: Spleen tyrosine kinase, BTK: Bruton’s tyrosine kinase, PKC-β: Protein kinase C β type, MALT1: Mucosa-associated lymphoid tissue lymphoma 1, Bcl-10: B-cell lymphoma/leukemia 10, JAK1/2: Janus kinase 1/2, STAT3/6: Signal transducer and activator of transcription 3/6, IL-4/10: Interleukin 4/10.
The main differential diagnosis in a case of PCNS DLBCL includes secondary involvement by systemic DLBCL, Burkitt lymphoma (BL, primary versus secondary, will be discussed below), and high-grade B-cell lymphoma (HGBL, a terminology used in the revised fourth edition of WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues) [12][10]. Two categories of HGBL have been described: HGBL with MYC and BCL2 and/or BCL6 rearrangements, and HGBL, not otherwise specified (NOS) [12][10]. Because, to our knowledge, BCL2 rearrangement has not been reported in PCNS DLBCL [35[24][31],42], the type of HGBL that will be encountered in PCNSL will most likely be HGBL, NOS, or HGBL with MYC and BCL6 rearrangements. HGBL, NOS is a heterogenous group of mature B-cell lymphomas that share morphologic, immunophenotypic, and genetic features with DLBCL and BL [12][10]. Whereas they may have MYC gene rearrangements, they are negative for both MYC plus BCL2 and/or BCL6 rearrangements [12][10]. Large, mature B-cell lymphomas with blastoid (medium-sized cells with fine, powdery chromatin) morphology fall into this category (HGBL, NOS) [12][10]. They can resemble Burkitt lymphoma in morphology, displaying sheets of monomorphic cells with scattered mitotic figures and apoptotic debris, intermixed with tingible body macrophages and imparting a starry sky appearance, but show immunophenotype and molecular genetic findings that are incompatible with BL [12][10]. In the cases with blastoid morphology, two main differential diagnoses need to be ruled out, namely B-lymphoblastic lymphoma/leukemia (positive for TdT and/or CD34) and the blastoid variant of mantle cell lymphoma (positive for Bcl-1/cyclin D1 and/or SOX11). HGBL is a rare and relatively new entity and there is paucity of data regarding its behavior in the PCNSL setting. Whereas a hematopathologist may make an effort to distinguish DLBCL and HGBL entities and to derive a more precise diagnosis, the therapeutic implications could be highly variable depending upon the preference of treating oncologist and specimen/resource availability.
References
- Cote, T.R.; Manns, A.; Hardy, C.R.; Yellin, F.J.; Hartge, P. AIDS/Cancer Study Group Epidemiology of Brain Lymphoma Among People With or Without Acquired Immunodeficiency Syndrome. J. Natl. Cancer Inst. 1996, 88, 675–679. [Google Scholar] [CrossRef]
- Dandachi, D.; Ostrom, Q.T.; Chong, I.; Serpa, J.A.; Giordano, T.P.; Kruchko, C.; Barnholtz-Sloan, J.S.; Fowler, N.; Colen, R.R.; Morón, F.E. Primary central nervous system lymphoma in patients with and without HIV infection: A multicenter study and comparison with U.S national data. Cancer Causes Control. 2019, 30, 477–488. [Google Scholar] [CrossRef]
- Matinella, A.; Lanzafame, M.; Bonometti, M.A.; Gajofatto, A.; Concia, E.; Vento, S.; Monaco, S.; Ferrari, S. Neurological complications of HIV infection in pre-HAART and HAART era: A retrospective study. J. Neurol. 2015, 262, 1317–1327. [Google Scholar] [CrossRef]
- Gopal, S.; Patel, M.R.; Yanik, E.L.; Cole, S.R.; Achenbach, C.J.; Napravnik, S.; Burkholder, G.A.; Reid, E.G.; Rodriguez, B.; Deeks, S.G.; et al. Temporal Trends in Presentation and Survival for HIV-Associated Lymphoma in the Antiretroviral Therapy Era. J. Natl. Cancer Inst. 2013, 105, 1221–1229. [Google Scholar] [CrossRef]
- Bataille, B.; Delwail, V.; Menet, E.; Vandermarcq, P.; Ingrand, P.; Wager, M.; Guy, G.; Lapierre, F. Primary intracerebral malignant lymphoma: Report of 248 cases. J. Neurosurg. 2000, 92, 261–266. [Google Scholar] [CrossRef]
- Grommes, C.; Rubenstein, J.L.; DeAngelis, L.M.; Ferreri, A.J.M.; Batchelor, T.T. Comprehensive approach to diagnosis and treatment of newly diagnosed primary CNS lymphoma. Neuro Oncol. 2019, 21, 296–305. [Google Scholar] [CrossRef]
- Grisariu, S.; Avni, B.; Batchelor, T.T.; Bent, M.J.V.D.; Bokstein, F.; Schiff, D.; Kuittinen, O.; Chamberlain, M.C.; Roth, P.; Nemets, A.; et al. Neurolymphomatosis: An International Primary CNS Lymphoma Collaborative Group report. Blood 2010, 115, 5005–5011. [Google Scholar] [CrossRef]
- Grimm, S.A.; Pulido, J.S.; Jahnke, K.; Schiff, D.; Hall, A.J.; Shenkier, T.N.; Siegal, T.; Doolittle, N.D.; Batchelor, T.T.; Herrlinger, U.; et al. Primary intraocular lymphoma: An International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann. Oncol. 2007, 18, 1851–1855. [Google Scholar] [CrossRef]
- Grimm, S.A.; Pulido, J.S.; Jahnke, K.; Schiff, D.; Hall, A.J.; Shenkier, T.N.; Siegal, T.; Doolittle, N.D.; Batchelor, T.T.; Herrlinger, U.; et al. Primary intraocular lymphoma: An International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann. Oncol. 2007, 18, 1851–1855. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. (Eds.) WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues, Revised, 4th ed.; IARC Press: Lyon, France, 2017. [Google Scholar]
- Abrey, L.E.; Batchelor, T.T.; Ferreri, A.J.; Gospodarowicz, M.; Pulczynski, E.J.; Zucca, E.; Smith, J.R.; Korfel, A.; Soussain, C.; DeAngelis, L.M.; et al. Report of an International Workshop to Standardize Baseline Evaluation and Response Criteria for Primary CNS Lymphoma. J. Clin. Oncol. 2005, 23, 5034–5043. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. (Eds.) WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues, Revised, 4th ed.; IARC Press: Lyon, France, 2017. [Google Scholar]
- Paulus, W.; Jellinger, K.; Hallas, C.; Ott, G.; Müller-Hermelink, H.K. Human herpesvirus-6 and Epstein-Barr virus genome in primary cerebral lymphomas. Neurology 1993, 43, 1591–1593. [Google Scholar] [CrossRef]
- Deckert, M.; Hans, V.H.; Eis-Hübinger, A.M.; Prinz, M.; Schaller, C.; Van Roost, D.; Aguzzi, A.; Wiestler, O.D.; Deckert, M. Human herpes virus-8 is not associated with primary central nervous system lymphoma in HIV-negative patients. Acta Neuropathol. 2001, 102, 489–495. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Besleaga, R.; Heinsohn, S.; Siebert, R.; Kabisch, H.; Wiestler, O.D.; Deckert, M. Absence of simian virus 40 DNA sequences in primary central nervous system lymphoma in HIV-negative patients. Virchows Archiv 2004, 444, 436–438. [Google Scholar] [CrossRef]
- Murray, J.; Morgello, S. Polyomaviruses and primary central nervous system lymphomas. Neurology 2004, 63, 1299–1301. [Google Scholar] [CrossRef]
- Ponzoni, M.; Berger, F.; Chassagne-Clement, C.; Tinguely, M.; Jouvet, A.; Ferreri, A.J.M.; Dell’Oro, S.; Terreni, M.R.; Doglioni, C.; Weis, J.; et al. Reactive perivascular T-cell infiltrate predicts survival in primary central nervous system B-cell lymphomas. Br. J. Haematol. 2007, 138, 316–323. [Google Scholar] [CrossRef]
- Makino, K.; Nakamura, H.; Shinojima, N.; Kuroda, J.-I.; Yano, S.; Mikami, Y.; Mukasa, A. BCL2 expression is associated with a poor prognosis independent of cellular origin in primary central nervous system diffuse large B-cell lymphoma. J. Neuro-Oncol. 2018, 140, 115–121. [Google Scholar] [CrossRef]
- Camilleri-Broët, S.; Crinière, E.; Broët, P.; Delwail, V.; Mokhtari, K.; Moreau, A.; Kujas, M.; Raphaël, M.; Iraqi, W.; Sautès-Fridman, C.; et al. A uniform activated B-cell–like immunophenotype might explain the poor prognosis of primary central nervous system lymphomas: Analysis of 83 cases. Blood 2005, 107, 190–196. [Google Scholar] [CrossRef]
- Lin, C.-H.; Kuo, K.; Chuang, S.-S.; Kuo, S.-H.; Chang, J.H.; Chang, K.-C.; Hsu, H.-C.; Tien, H.; Cheng, A.-L. Comparison of the Expression and Prognostic Significance of Differentiation Markers between Diffuse Large B-Cell Lymphoma of Central Nervous System Origin and Peripheral Nodal Origin. Clin. Cancer Res. 2006, 12, 1152–1156. [Google Scholar] [CrossRef]
- Hattab, E.M.; Martin, S.E.; Al-Khatib, S.M.; Kupsky, W.J.; Vance, G.H.; Stohler, R.A.; Czader, M.; Al-Abbadi, M.A. Most primary central nervous system diffuse large B-cell lymphomas occurring in immunocompetent individuals belong to the nongerminal center subtype: A retrospective analysis of 31 cases. Mod. Pathol. 2010, 23, 235–243. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Schmitz, R.; Courts, C.; Stenzel, W.; Bechtel, D.; Niedobitek, G.; Blumcke, I.; Reifenberger, G.; Von Deimling, A.; Jungnickel, B.; et al. Absence of Immunoglobulin Class Switch in Primary Lymphomas of the Central Nervous System. Am. J. Pathol. 2005, 166, 1773–1779. [Google Scholar] [CrossRef]
- Brunn, A.; Nagel, I.; Montesinos-Rongen, M.; Klapper, W.; Vater, I.; Paulus, W.; Hans, V.; Blümcke, I.; Weis, J.; Siebert, R.; et al. Frequent triple-hit expression of MYC, BCL2, and BCL6 in primary lymphoma of the central nervous system and absence of a favorable MYClowBCL2low subgroup may underlie the inferior prognosis as compared to systemic diffuse large B cell lymphomas. Acta Neuropathol. 2013, 126, 603–605. [Google Scholar] [CrossRef]
- Gill, K.Z.; Iwamoto, F.; Allen, A.; Hoehn, D.; Murty, V.V.; Alobeid, B.; Bhagat, G. MYC Protein Expression in Primary Diffuse Large B-Cell Lymphoma of the Central Nervous System. PLoS ONE 2014, 9, e114398. [Google Scholar] [CrossRef]
- Cady, F.M.; O’Neill, B.P.; Law, M.E.; Decker, P.A.; Kurtz, D.M.; Giannini, C.; Porter, A.B.; Kurtin, P.J.; Johnston, P.B.; Dogan, A.; et al. Del(6)(q22) and BCL6 Rearrangements in Primary CNS Lymphoma Are Indicators of an Aggressive Clinical Course. J. Clin. Oncol. 2008, 26, 4814–4819. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Zühlke-Jenisch, R.; Gesk, S.; Martín-Subero, J.I.; Schaller, C.; Van Roost, D.; Wiestler, O.D.; Deckert, M.; Siebert, R. Interphase Cytogenetic Analysis of Lymphoma-Associated Chromosomal Breakpoints in Primary Diffuse Large B-Cell Lymphomas of the Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 926–933. [Google Scholar] [CrossRef]
- Shi, Q.-Y.; Feng, X.; Bao, W.; Ma, J.; Lv, J.-H.; Wang, X.; Rao, Q.; Shi, Q.-L. MYC/BCL2 Co-Expression Is a Stronger Prognostic Factor Compared With the Cell-of-Origin Classification in Primary CNS DLBCL. J. Neuropathol. Exp. Neurol. 2017, 76, 942–948. [Google Scholar] [CrossRef]
- Kim, S.; Nam, S.J.; Kwon, D.; Kim, H.; Lee, E.; Kim, T.M.; Heo, D.S.; Park, S.H.; Kim, C.W.; Jeon, Y.K. MYC and BCL2 overexpression is associated with a higher class of Memorial Sloan-Kettering Cancer Center prognostic model and poor clinical outcome in primary diffuse large B-cell lymphoma of the central nervous system. BMC Cancer 2016, 16, 1–11. [Google Scholar] [CrossRef]
- Tapia, G.; Baptista, M.-J.; Muñoz-Marmol, A.-M.; Gaafar, A.; Puente-Pomposo, M.; Garcia, O.; Marginet-Flinch, R.; Sanz, C.; Navarro, J.-T.; Sancho, J.-M.; et al. MYC protein expression is associated with poor prognosis in primary diffuse large B-cell lymphoma of the central nervous system. APMIS 2015, 123, 596–603. [Google Scholar] [CrossRef]
- Fukumura, K.; Kawazu, M.; Kojima, S.; Ueno, T.; Sai, E.; Soda, M.; Ueda, H.; Yasuda, T.; Yamaguchi, H.; Lee, J.; et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol. 2016, 131, 865–875. [Google Scholar] [CrossRef]
- Son, S.-M.; Ha, S.-Y.; Yoo, H.-Y.; Oh, D.; Kim, S.J.; Kim, W.-S.; Ko, Y.-H. Prognostic impact of MYC protein expression in central nervous system diffuse large B-cell lymphoma: Comparison with MYC rearrangement and MYC mRNA expression. Mod. Pathol. 2016, 30, 4–14. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Liu, Y.; Liu, Z.; Cui, Q.; Ji, N.; Sun, S.; Wang, B.; Wang, Y.; Sun, X.; et al. Immunohistochemical profile and prognostic significance in primary central nervous system lymphoma: Analysis of 89 cases. Oncol. Lett. 2017, 14, 5505–5512. [Google Scholar] [CrossRef]
- Guo, S.; Bai, Q.; Rohr, J.M.; Wang, Y.; Liu, Y.; Zeng, K.; Yu, K.; Zhang, X.; Wang, Z. Clinicopathological features of primary diffuse large B-cell lymphoma of the central nervous system-strong EZH2 expression implying diagnostic and therapeutic implication. APMIS 2016, 124, 1054–1062. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Küppers, R.; Schlüter, D.; Spieker, T.; Van Roost, D.; Schaller, C.; Reifenberger, G.; Wiestler, O.D.; Deckert-Schlüter, M. Primary Central Nervous System Lymphomas Are Derived from Germinal-Center B Cells and Show a Preferential Usage of the V4–34 Gene Segment. Am. J. Pathol. 1999, 155, 2077–2086. [Google Scholar] [CrossRef]
- Sharathkumar, B.; Jon, D.W. Primary Central Nervous System Lymphoma. Arch. Pathol. Lab. Med. 2008, 132, 1830–1834. [Google Scholar]
- Rubenstein, J.L.; Fridlyand, J.; Shen, A.; Aldape, K.; Ginzinger, D.; Batchelor, T.; Treseler, P.; Berger, M.; McDermott, M.; Prados, M.; et al. Gene expression and angiotropism in primary CNS lymphoma. Blood 2006, 107, 3716–3723. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Godlewska, E.; Brunn, A.; Wiestler, O.D.; Siebert, R.; Deckert, M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011, 122, 791–792. [Google Scholar] [CrossRef]
- Schwindt, H.; Vater, I.; Kreuz, M.; Montesinos-Rongen, M.; Brunn, A.; Richter, J.; Gesk, S.; Ammerpohl, O.; Wiestler, O.D.; Hasenclever, D.; et al. Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia 2009, 23, 1875–1884. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]