Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Fanny Huang and Version 1 by Danila Coradini.
Cholesterol (CHOL) is a multifaceted lipid molecule. It is an essential structural component of cell membranes, where it cooperates in regulating the intracellular trafficking and signaling pathways. Additionally, it serves as a precursor for vital biomolecules, including steroid hormones, isoprenoids, vitamin D, and bile acids. Although CHOL is normally uptaken from the bloodstream, cells can synthesize it de novo in response to an increased requirement due to physiological tissue remodeling or abnormal proliferation, such as in cancer. Cumulating evidence indicated that increased CHOL biosynthesis is a common feature of breast cancer and is associated with the neoplastic transformation of normal mammary epithelial cells.
- cholesterol biosynthesis
- Hippo signaling pathway
- invasive breast cancer
1. Introduction
Cholesterol (CHOL) is the most common sterol of vertebrates, and its homeostasis is a dynamic balance between absorption and de novo synthesis. While higher CHOL intake from food leads to a net decreased endogenous production, the body compensates for the poorly absorbed neutral cholesteryl esters by synthesizing de novo CHOL primarily in the liver [1]. Once synthesized, CHOL is transported through the bloodstream around the body, packaged within lipoprotein particles, mainly low-density lipoproteins (LDLs), which can carry up to 6000 fat molecules per particle [2].
Cellular CHOL homeostasis reflects this dynamic balance. Specific receptors (LDLRs), located on the plasma membrane, facilitate the uptake of LDLs, and once internalized, they are stored in endosomes, where they undergo lipase-dependent hydrolysis, making CHOL available for various cellular processes [3]. Through a feedback regulatory mechanism, this exogenously produced CHOL inhibits the de novo synthesis by repressing HMGCR, the gene coding for 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase, the enzyme catalyzing the first rate-limiting step in the biosynthetic process (Figure 1).
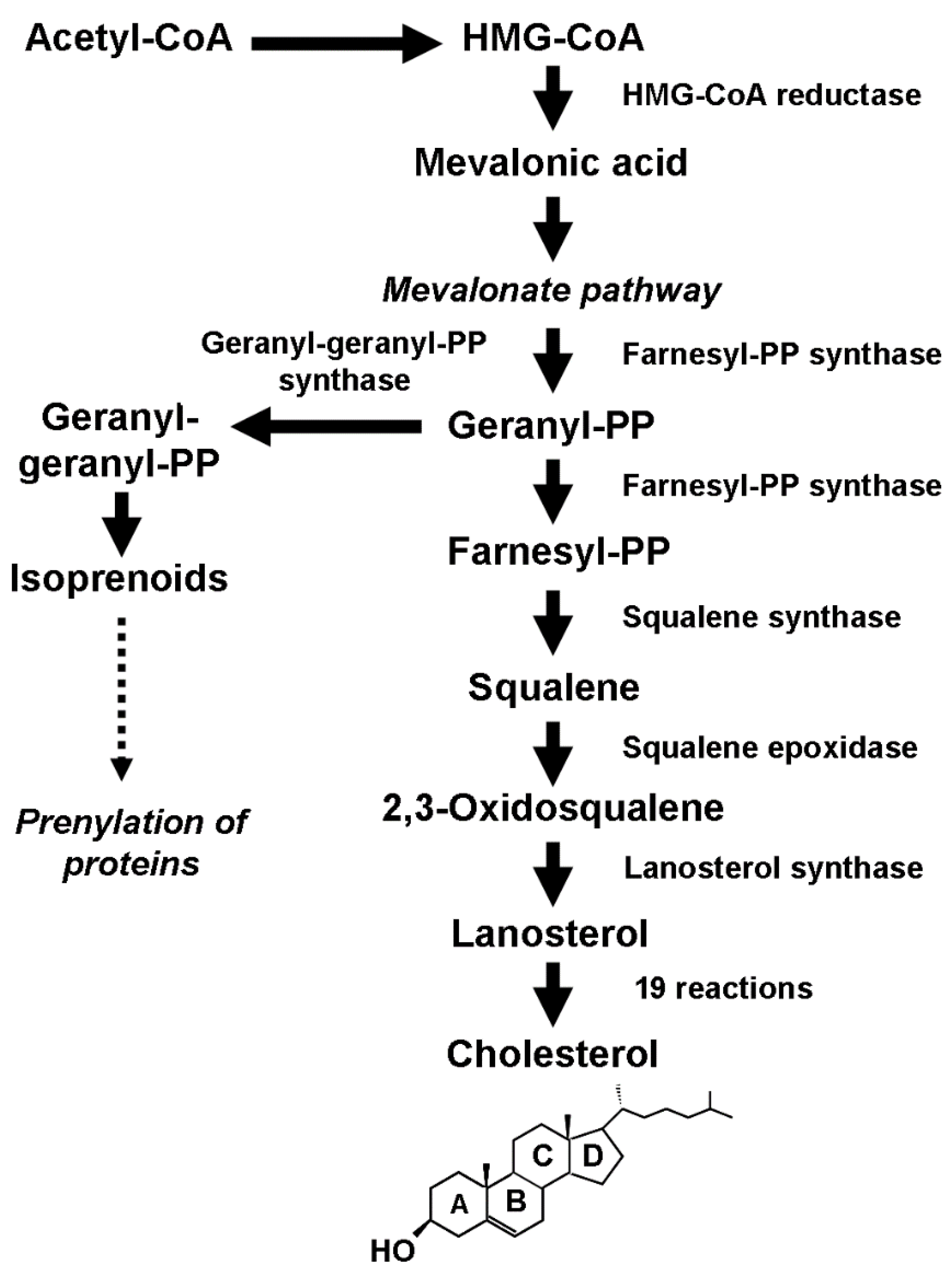
Figure 1.
Schematic description of the biosynthetic process that leads to the production of cholesterol and isoprenoids.
The transcription of HMGCR is governed by sterol regulatory element-binding protein (SREBP) transcription factors, which are sensitive to CHOL concentration. In the presence of CHOL, SREBP precursors are retained in the membrane of the endoplasmic reticulum by a tight association with a multi-pass membrane protein called SREBP cleavage-activating protein (SCAP), which has a sterol-sensing domain to which CHOL can bind [4]. SCAP also forms a reversible ternary complex with another multi-pass membrane protein called the insulin-induced gene (INSIG), which promotes the ubiquitin-mediated degradation of HMG-CoA reductase by recruiting the membrane-associated ubiquitin ligase gp78 [5,6,7][5][6][7].
Conversely, under CHOL deprivation conditions, SCAP and INSIG no longer bind. INSIG is degraded, whereas SCAP undergoes a conformational change in the sterol-sensing domain that unmasks the endoplasmic reticulum export signal. The SCAP/SREBP complex is then packed into COPII-coated vesicles and targeted to the Golgi apparatus, where SREBP is sequentially cleaved by site-1 protease and site-2 protease (S1P and S2P) [5]. The N-terminal domain of SREBP resulting from cleavage can enter the nucleus, bind to sterol-responsive elements (SREs), and activate the transcription of both the gene coding for the LDL receptor to enhance the intracellular CHOL uptake and those coding for HMG-CoA reductase and squalene epoxidase (SQLE), the enzyme that catalyzes the irreversible commitment to CHOL, to increase the endogenous production.
32. Cholesterol Biosynthesis and Breast Cancer Initiation and Development
Elevated levels of circulating CHOL due to CHOL homeostasis dysregulation can cause severe metabolic dysfunctions. High serum levels of CHOL are a well-known risk factor for heart disease [55][8], and cumulating evidence suggests that hypercholesterolemia may also be associated with the risk of developing several types of tumors, including breast cancer [56,57,58][9][10][11]. However, observational studies on the relationship between circulating CHOL and breast cancer risk have provided inconclusive or sometimes contradictory results. While some systematic reviews and meta-analyses suggest that dietary CHOL intake and serum CHOL levels are associated with breast cancer risk [59[12][13],60], other prospective studies have found no significant or inverse association [61,62,63][14][15][16]. Confounding variables such as obesity, menopausal status, tumor subtype, and ER status may be the reason for these conflicting results.
Conversely, preclinical studies have provided compelling evidence that tumors show a CHOL concentration higher than normal tissue, indicating a significant change in CHOL homeostasis [8][17]. This finding agrees with the increased requirement for CHOL of transformed cells to sustain their abnormal proliferation. They satisfy this requirement by increasing the uptake of exogenous CHOL through the overexpression of LDL receptors and the endogenous production through de novo biosynthesis. Recent findings have also shown that the dysregulation of the mevalonate pathway, the core of the biosynthetic process (Figure 1), results in the overproduction of isoprenoids [64][18] that play an essential role in the posttranslational modification (prenylation) of several proteins, including Rho GTPases (Rac, RhoA, Cdc42). These proteins act as molecular switches in various cell processes and signaling pathways, including the Hippo signaling pathway.
3.1. Cholesterol Biosynthesis and the Hippo Signaling Pathway
2.1. Cholesterol Biosynthesis and the Hippo Signaling Pathway
First discovered in Drosophila, the evolutionarily conserved Hippo signaling pathway limits organ size by controlling tissue growth and preventing the uncontrolled proliferation of progenitor cells [65][19]. The core of the Hippo pathway consists of two highly conserved serine/threonine kinases called serine/threonine kinase 3/4 (STK3/4), also known as mammalian Ste20-like kinase 1/2 (MST1/2), and large tumor suppressor 1/2 (LATS1/2). When cells stop growth in response to cell contact inhibition, these kinases phosphorylate the Yes-associated protein 1 (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) coactivators [66][20], preventing them from entering the nucleus and promoting their degradation.
As schematically depicted in Figure 2, the signaling cascade starts when MTS1/2 associates with the scaffold protein Salvador family WW domain-containing protein 1 (SAV1) to phosphorylate and activate LATS1/2. Once activated, LATS1/2 associates with the regulatory MOB kinase activator 1/2 (MOB1/2) and phosphorylates the YAP/TAZ coactivator on multiple sites, including Ser168, which functions as the binding site for 14-3-3 protein. The latter sequesters YAP/TAZ in the cytoplasm and primes for its rapid proteasomal degradation, thereby inhibiting their migration into the nucleus [67][21]. Conversely, when YAP/TAZ coactivators are not phosphorylated, they can enter the nucleus, associate with TEA domain transcription factor 1/4 (TEAD1/4), and regulate the expression of several genes involved in cell proliferation promotion and inhibition of apoptosis.
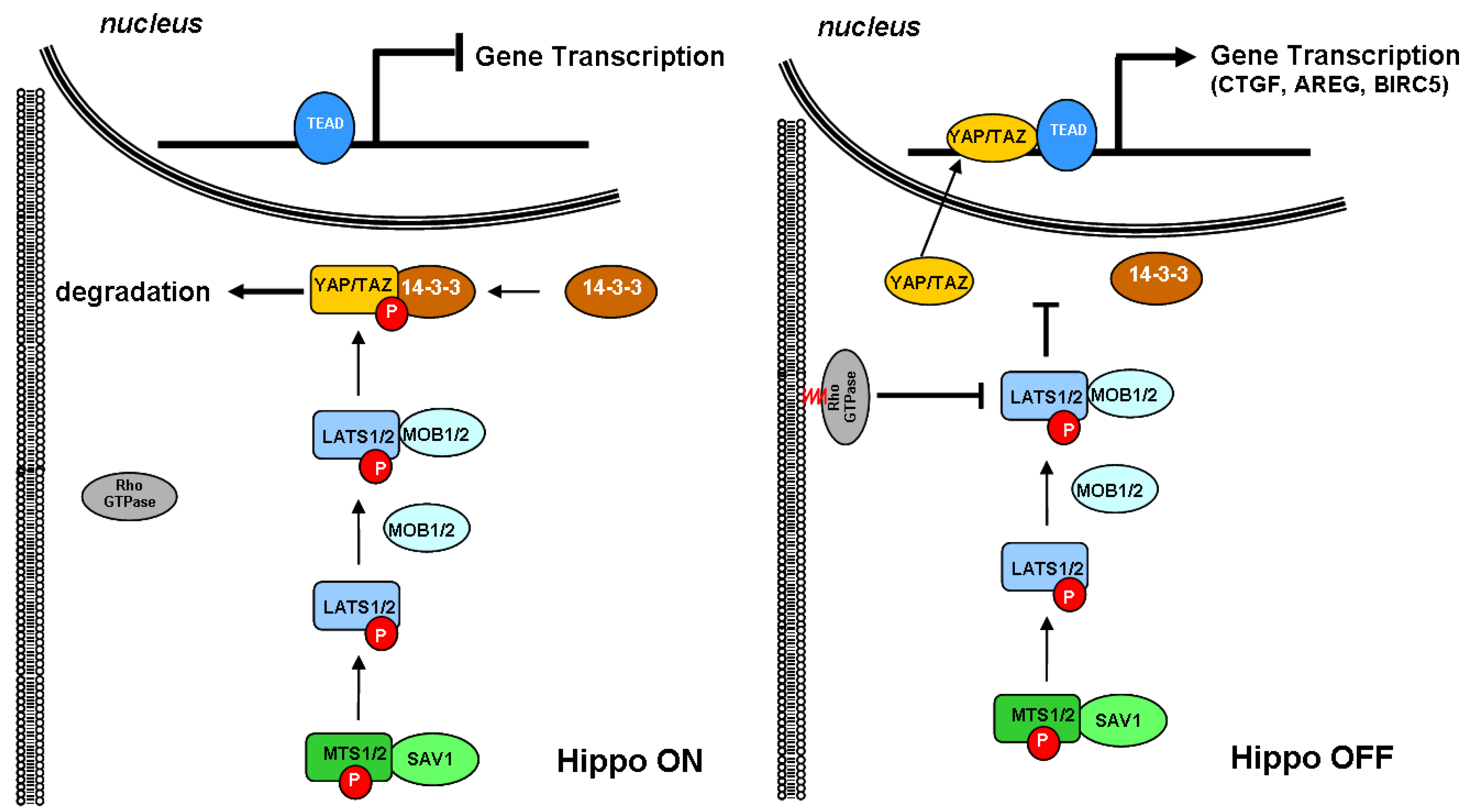
Figure 2. Schematic depiction of the interaction between small GTPase and Hippo signaling pathways. The core of the Hippo pathway consists of a kinase cascade where STK3, in complex with its regulatory protein SAV1, phosphorylates and activates LATS1. After forming a complex with the regulatory protein MOB1A, LATS1 phosphorylates and inactivates YAP. Phosphorylation of YAP by LATS1 inhibits its translocation into the nucleus, the binding to TEAD, and the transcription of several genes involved in cell proliferation, apoptosis, and migration. TEAD, TEA domain transcription factor; YAP, Yes-associated protein; TAZ, transcriptional coactivator with PDZ-binding motif; LATS1, large tumor suppressor kinase 1; MOB1A, MOB kinase activator 1A; STK3, serine/threonine kinase 3; SAV1, Salvador family WW domain-containing protein 1.
Cumulating evidence indicated that Rho GTPases play a crucial role in promoting the nuclear translocation of YAP/TAZ. Rho GTPases are usually found free in the cytoplasm. However, when structurally modified by the enzymatic activity of the geranylgeranyl pyrophosphate produced by the mevalonate cascade, they anchor to cell membranes and act as a transducer of the changes occurring in the cytoskeleton dynamics. In response to growth factors, Rho GTPases coordinate the rearrangement of the actin stress fibers, which, in turn, sequester angiomotin, an inhibitor of YAP/TAZ, and promote the nuclear translocation of the coactivators [68,69][22][23]. Previously considered independent, the Rho GTPase-mediated activation of YAP/TAZ is now emerging as linked to the Hippo pathway because of the direct inhibition of LATS1/2 kinase [70][24].
Given the role played by the Hippo signaling pathway in controlling the growth of normal cells, it is not surprising that the disruption of this pathway leads to the constitutive activation of YAP/TAZ, which in turn promotes uncontrolled proliferation and tumorigenesis [66,71][20][25], and the finding that common solid tumors, including breast cancer, show a high expression of YAP protein [72][26].
3.2. Cholesterol Biosynthesis, Mammary Stem Cells, and Breast Cancer Stem Cells
2.2. Cholesterol Biosynthesis, Mammary Stem Cells, and Breast Cancer Stem Cells
The mammary gland is a highly dynamic organ that undergoes profound changes throughout the woman’s reproductive life. Starting at puberty and until menopause, these changes are more noticeable during pregnancy and lactation when the glandular tissue proliferates, undergoes complete differentiation, and secretes milk due to the influence of sex and lactogenic hormones.
The functional structure of the human mammary gland consists of 6–12 independent ductal–lobular systems drained by collecting ducts that converge at the nipple (Figure 3). Each ductal-lobular system contains several terminal ductal–lobular units (TDLUs) that represent the secretory unit of the mammary gland. TDLUs contain lobules or alveoli drained by a duct. The alveoli are composed of lobular epithelial cells, while the ducts are composed of epithelial cells (facing the lumen) and myoepithelial cells that form the basal layer. TDLUs are surrounded by stroma, which contains different kinds of cells such as adipocytes, endothelial cells, immune cells, and fibroblasts, all of which are embedded in the extracellular matrix.
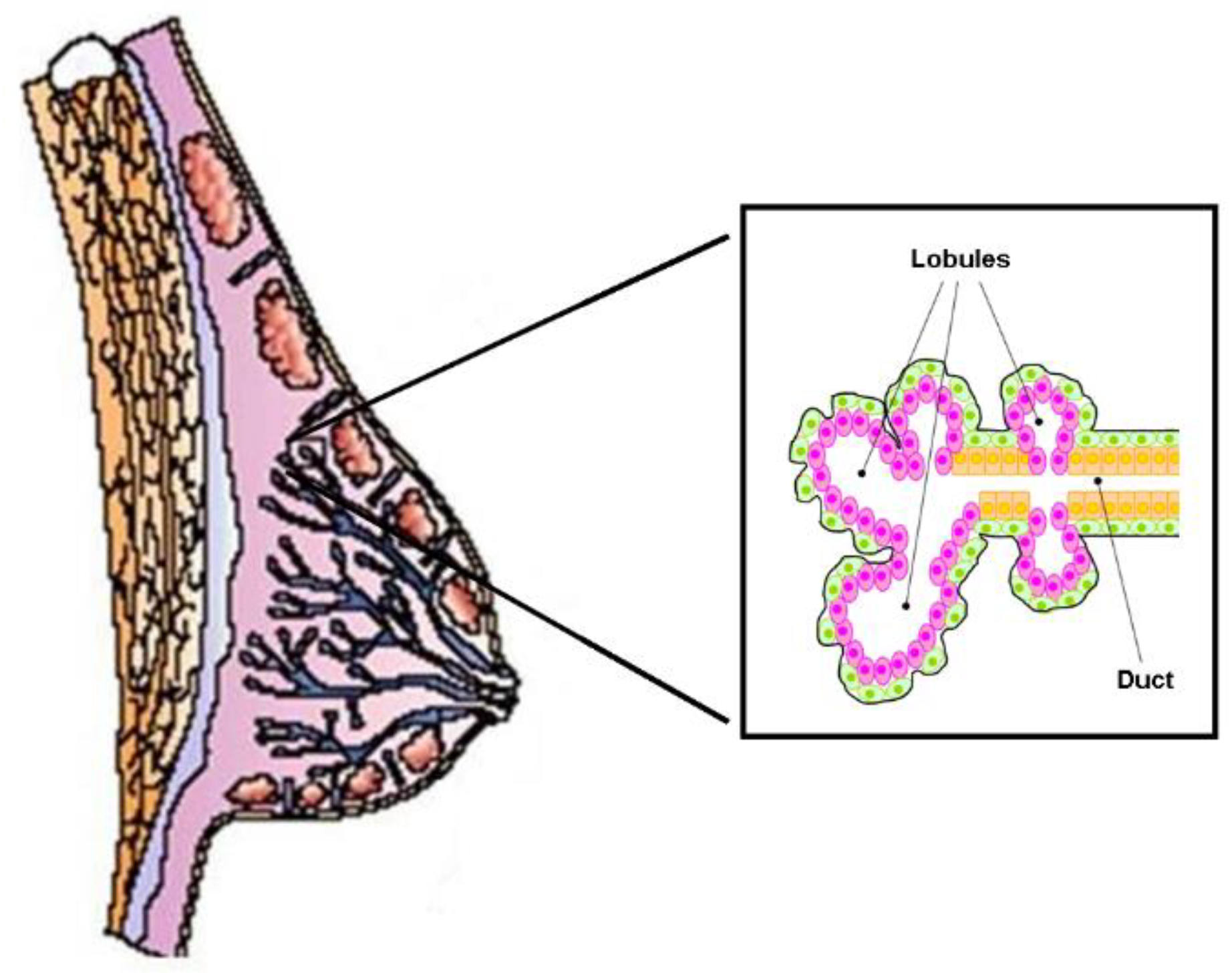
Figure 3. Schematic structure of the normal mammary gland. It consists of independent duct lobular systems drained by collecting ducts that converge at the nipple. The inset shows the cellular components of the terminal ductal–lobular unit (TDLU), the basic secretory unit of the mammary gland. Lobular (pink) and ductal (orange) epithelial cells are separated from the surrounding stroma by myoepithelial elements (in green).
Epithelial and myoepithelial cells derive from common ancestors known as multipotent mammary epithelial stem cells (MaSCs), which have the unique capacity to self-renew and to generate the lineage-restricted progenitors of both lobular and ductal epithelial cells as well as myoepithelial cells [73,74,75][27][28][29]. These progenitors, located in distinct areas of the developing mammary known as niches, proliferate extensively and differentiate to form TDLUs under the control of ovarian hormones [76,77][30][31].
Since the discovery of MaSC, significant progress has been made in identifying the pathways that control the self-renewal, the lineage commitment, and the differentiation of stem and progenitor cells during the development of the normal mammary gland and their potential involvement in cancer initiation [78,79][32][33]. There is now cumulating evidence that the signaling pathways that regulate the physiological turnover of the mammary tissue are often disrupted in breast cancer, suggesting that stem and progenitor cells, due to critical gene mutations and epigenetic events, can give rise to a small population of highly tumorigenic cells known as breast cancer stem cells (BCSCs) [80,81,82,83][34][35][36][37].
Recent preclinical studies have found that CSC-enriched breast cancer cell lines have an increased expression of the enzymes involved in the de novo synthesis of CHOL compared to control cells [84][38]. This finding suggests that CHOL biosynthesis plays a crucial role in BCSC propagation and tumor development, especially considering the link between estradiol, the most potent bioactive CHOL derivative, and breast cancer growth. Indeed, estradiol promotes and fuels the proliferation of estrogen-dependent progenitors and contributes to the development of ER-positive tumors that account for about 70% of breast cancer diagnoses.
Additionally, studies have shown that the mevalonate metabolic pathway is remarkably hyperactivated in CSC-enriched breast cancer cell lines. However, this hyperactivation was reduced by treatment with statins, leading to decreased isoprenoid production and less availability of anchorage sites to the membrane for the Rho GTPases [85][39]. This finding suggests that the dysregulation of CHOL biosynthesis, which leads to the over-production of isoprenoids, may modify the physiological turnover of stem and progenitor cells, promoting their transformation and proliferation.
Interestingly, the downregulation of the mevalonate precursor enzyme, HMG-CoA synthase (HMGCS1), through silencing the expression of the corresponding gene, resulted in a significant decrease in BCSC fraction. This finding proposes HMGCS1 as a potential gatekeeper for the mevalonate pathway metabolism, whose dysregulation enhances cancer stem cell enrichment [86][40]. Blocking the expansion of BCSCs through inhibition of the HMGCS1 gene could be a more efficient approach to control the over-production of isoprenoids compared to standard doses of statins, thereby reducing the proliferation of progenitor cells and cancer development.
Another promising approach to prevent the expansion of BCSCs by regulating CHOL biosynthesis involves microRNAs (miRNAs). They are small (~18–25 nucleotides) non-encoding RNAs that can modulate the expression of the genes involved in the pathway [87][41]. A recent study using bioinformatics analysis [88][42] has revealed that hsa-miR-34a-5p and hsa-miR-373-3p can affect the expression of several genes related to CHOL, including INSIG2. This gene codes for insulin-induced gene 2, an endoplasmic reticulum protein that acts as a negative regulator of CHOL biosynthesis by retaining the SCAP-SREBP complex in the endoplasmic reticulum. As a result, SREBP processing was blocked, and the transcription of the target genes, including HMGCS1, was inhibited.
3.3. Cholesterol Biosynthesis and Ductal Carcinoma In Situ
2.3. Cholesterol Biosynthesis and Ductal Carcinoma In Situ
Ductal carcinoma in situ (DCIS) is a non-invasive form of breast cancer whose incidence has risen markedly since the early 1980s due to the widespread use of mammography screening. According to the American Cancer Society, in 2022, DCIS accounted for 15% of all new mammographically detected breast cancers, 82% of which occurred in women aged 50 years and older [89][43]. Although DCIS is considered a non-obligate precursor to invasive breast cancer (IBC), if left untreated, about 50% of cases progress to IBC, even several decades after diagnosis. A 30-year follow-up study showed that 36% of untreated low-grade DCIS progressed to IBC [90][44].
While many efforts have been made to elucidate the molecular changes associated with DCIS progression [91[45][46][47],92,93], less attention has been given to the molecular and metabolic changes that lead to the transformation of the normal mammary epithelium in DCIS. Identifying the early molecular events that trigger and drive this transformation is essential to prevent DCIS with appropriate and effective treatments.
In a recent study, researchers investigated the expression of some genes involved in CHOL biosynthesis in a set of patient-matched samples of DCIS and corresponding histologically normal (HN) epithelium [94][48]. Researchers found that HMGCR, which encodes the enzyme that controls the rate-limiting step in CHOL biosynthesis, was more expressed in the epithelial compartment of DCIS than in the corresponding normal epithelium. This finding agrees with the clinical evidence that the HMG-CoA reductase (evaluated as immunohistochemical cytoplasmic staining) is moderately/strongly expressed in about 70% of the assessed DCIS samples [95][49]. Similarly, some crucial downstream genes involved in CHOL (FDFT1, SQLE, and LSS) and isoprenoid production (GGPS1) were more expressed in DCIS.
Researchers also found that HMGCS1, the gene coding for the enzyme upstream to the mevalonate pathway, was more expressed in DCIS than in the corresponding normal tissue. This result supports the hypothesis that DCIS may originate from transformed progenitor cells, where the overexpression of HMG-CoA synthase plays a crucial role in dysregulating the biosynthetic process.
Apart from the differential expression, the genes involved in CHOL biosynthesis changed their interrelationship dramatically in DCIS. Thus, for example, the positive association between HMGCR and the downstream GGPS1, observed in normal tissue, disappeared in DCIS, and the positive association between GGPS1 and SQLE, which regulate, respectively, the production of isoprenoids and the irreversible commitment to CHOL [96][50], switched to a negative one. The result suggests that, due to the dysregulation of the biosynthetic process, CHOL production was overcome by that of isoprenoids.
Collectively, these results support the hypothesis of a link between the dysregulation of the mevalonate pathway and the transcriptional activity of the YAP/TAZ coactivator through the increased production of the isoprenoids required for the prenylation of small GTPases, followed by GTPases anchoring to the cell membrane and activation, inhibition of the Hippo signaling pathway and promotion of YAP/TAZ coactivator nuclear translocation. Moreover, these results substantiate the use of CHOL-lowering drugs to restore the correct equilibrium in the production of isoprenoids and CHOL, thereby blocking cell proliferation and preventing DCIS progression to a more aggressive form.
3.4. Cholesterol Biosynthesis and Invasive Breast Cancer
2.4. Cholesterol Biosynthesis and Invasive Breast Cancer
Considering the natural history of breast cancer, it is not surprising that CHOL biosynthesis is dysregulated in IBC, where CHOL and its derivatives contribute to cancer cell proliferation and dissemination [97][51]. Preclinical studies have shown that CHOL not only triggers metabolic switching through the activation of the ERRα pathway but also contributes to cancer cell invasion by increasing tumor sphere formation [98][52] and the number of the cholesterol-enriched rafts [99][53], where the membrane-associated receptors for growth factors, adhesion molecules, and immunoregulatory molecules [100][54] are concentrated.
In a recent study aimed at exploring the molecular basis of CHOL biosynthesis dysregulation in paired samples of IBC and adjacent HN tissue [101][55], it was found that the majority of the genes coding for the enzymes involved in CHOL biosynthesis were more expressed in tumors than the corresponding HN tissues. In particular, the genes differentially expressed code for the enzymes involved in rate-limiting or commitment steps of the biosynthetic process: HMG-CoA synthase (coded by HMGCS1), is recognized as the gatekeeper of the pathway, HMG-CoA reductase (coded by HMGCR), is known as the first rate-limiting enzyme, squalene monooxygenase (coded by SQLE), is the first committed step of cholesterol biosynthesis, and NSDHL that codes for the NAD(P)-dependent steroid dehydrogenase-like, has recently been associated with an unfavorable prognosis and metastatic spread [102][56].
The study also found that the expression level of HMGCS1, HMGCR, SQLE, and NSDHL progressively increased with tumor grade and correlated positively with the expression of MKI67, the gene coding for the proliferation marker Ki67. The association between the dysregulated CHOL biosynthesis and tumor aggressiveness was confirmed by the inverse association between the tumor expression level of HMGCR or NSDHL and the patient’s relapse-free survival. Even after taking into account tumor grade and MKI67 expression level as confounding covariates, patients with high expression levels of both genes had a significantly higher risk of relapse than those with low expression levels.
The finding that the expression profile of the genes involved in de novo CHOL biosynthesis progressively changed with tumor grade is particularly significant, considering that 40–60% of the diagnosed breast cancers fall in the heterogeneous class of G2 tumors which are characterized by a highly variable morphology, unpredictable risk of distant metastasis recurrence, [103][57] and not always adequate treatment [104][58]. Therefore, evaluating the expression profile of this panel of genes could significantly improve the classification of G2 tumors and help select the most effective therapeutic approach to improve the patient’s prognosis.
References
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245.
- Sugii, S.; Reid, P.C.; Ohgami, N.; Du, H.; Chang, T.Y. Distinct Endosomal Compartments in Early Trafficking of Low Density Lipoprotein-Derived Cholesterol. J. Biol. Chem. 2003, 278, 27180–27189.
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol Sensing, Trafficking, and Esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157.
- Gao, Y.; Zhou, Y.; Goldstein, J.L.; Brown, M.S.; Radhakrishnan, A. Cholesterol-Induced Conformational Changes in the Sterol-Sensing Domain of the Scap Protein Suggest Feedback Mechanism to Control Cholesterol Synthesis. J. Biol. Chem. 2017, 292, 8729–8737.
- Sun, L.P.; Li, L.; Goldstein, J.L.; Brown, M.S. Insig Required for Sterol-Mediated Inhibition of Scap/SREBP Binding to COPII Proteins in vitro. J. Biol. Chem. 2005, 280, 26483–26490.
- Sever, N.; Yang, T.; Brown, M.S.; Goldstein, J.L.; DeBose-Boyd, R.A. Accelerated Degradation of HMG CoA Reductase Mediated by Binding of Insig-1 to Its Sterol-Sensing Domain. Mol. Cell. 2003, 11, 25–33.
- DeBose-Boyd, R.A. Feedback Regulation of Cholesterol Synthesis: Sterol-Accelerated Ubiquitination and Degradation of HMG CoA Reductase. Cell Res. 2008, 18, 609–621.
- Clark, L.T. Cholesterol and Heart Disease: Current Concepts in Pathogenesis and Treatment. J. Natl. Med. Assoc. 1986, 78, 743–751.
- Brown, A.J. Cholesterol, Statins and Cancer. Clin. Exp. Pharmacol. Physiol. 2007, 34, 135–141.
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070.
- Murdock, D.J.; Sanchez, R.J.; Mohammadi, K.A.; Fazio, S.; Geba, G.P. Serum Cholesterol and the Risk of Developing Hormonally Driven Cancers: A Narrative Review. Cancer Med. 2023, 12, 6722–6767.
- Li, C.; Yang, L.; Zhang, D.; Jiang, W. Systematic Review and Meta-Analysis Suggest that Dietary Cholesterol Intake Increases Risk of Breast Cancer. Nutr. Res. 2016, 36, 627–635.
- Johnson, K.E.; Siewert, K.M.; Klarin, D.; Damrauer, S.M.; Chang, K.M.; Tsao, P.S.; Assimes, T.L.; Maxwell, K.N.; Voight, B.F. The Relationship Between Circulating Lipids and Breast Cancer Risk: A Mendelian randomization study. PLoS Med. 2020, 17, e1003302.
- Touvier, M.; Fassier, P.; His, M.; Norat, T.; Chan, D.S.; Blacher, J.; Hercberg, S.; Galan, P.; Druesne-Pecollo, N.; Latino-Martel, P. Cholesterol and Breast Cancer Risk: A Systematic Review and Meta-Analysis of Prospective Studies. Br. J. Nutr. 2015, 114, 347–357.
- Baek, A.E.; Nelson, E.R. The Contribution of Cholesterol and Its Metabolites to the Pathophysiology of Breast Cancer. Horm. Cancer 2016, 7, 219–228.
- Ben Hassen, C.; Goupille, C.; Vigor, C.; Durand, T.; Guéraud, F.; Silvente-Poirot, S.; Poirot, M.; Frank, P.G. Is Cholesterol a Risk Factor for Breast Cancer Incidence and Outcome? J. Steroid Biochem. Mol. Biol. 2023, 232, 106346.
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of Cholesterol in the Development and Progression of Breast Cancer. Am. J. Pathol. 2011, 178, 402–412.
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the Mevalonate Pathway Promotes Transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056.
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo Pathway in Organ Size Control, Tissue Regeneration and Stem Cell Self-Renewal. Nat. Cell Biol. 2011, 13, 877–883.
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21, 2747–2761.
- Yu, F.X.; Guan, K.L. The Hippo Pathway: Regulators and Regulations. Genes Dev. 2013, 27, 355–371.
- Etienne-Manneville, S.; Hall, A. Rho GTPases in Cell Biology. Nature 2002, 420, 629–635.
- Ridley, A.J.; Hall, A. The Small GTP-Binding Protein Rho Regulates the Assembly of Focal Adhesions and Actin Stress Fibers in Response to Growth Factors. Cell 1992, 70, 389–399.
- Jang, J.W.; Kim, M.K.; Bae, S.C. Reciprocal Regulation of YAP/TAZ by the Hippo Pathway and the Small GTPase Pathway. Small GTPases 2020, 11, 280–288.
- Zhao, B.; Li, L.; Lei, Q.; and Guan, K.L. The Hippo-YAP Pathway in Organ Size Control and Tumorigenesis: An Updated Version. Genes Dev. 2010, 24, 862–874.
- Steinhardt, A.A.; Gayyed, M.F.; Klein, A.P.; Dong, J.; Maitra, A.; Pan, D.; Montgomery, E.A.; Anders, R.A. Expression of Yes-associated Protein in Common Solid Tumors. Hum. Pathol. 2008, 39, 1582–1589.
- Stingl, J.; Eaves, C.J.; Kuusk, U.; Emerman, J.T. Phenotypic and Functional Characterization In Vitro of a Multipotent Epithelial Cell Present in the Normal Adult Human Breast. Differentiation 1998, 63, 201–213.
- Gudjonsson, T.; Villadsen, R.; Nielsen, H.L.; Rønnov-Jessen, L.; Bissell, M.J.; Petersen, O.W. Isolation, Immortalization, and Characterization of a Human Breast Epithelial Cell Line with Stem Cell Properties. Genes Dev. 2002, 16, 693–706.
- Fu, N.Y.; Nolan, E.; Lindeman, G.J.; Visvader, J.E. Stem Cells and the Differentiation Hierarchy in Mammary Gland Development. Physiol. Rev. 2020, 100, 489–523.
- Joshi, P.A.; Jackson, H.W.; Beristain, A.G.; Di Grappa, M.A.; Mote, P.A.; Clarke, C.L.; Stingl, J.; Waterhouse, P.D.; Khokha, R. Progesterone Induces Adult Mammary Stem Cell Expansion. Nature 2010, 465, 803–807.
- Honeth, G.; Schiavinotto, T.; Vaggi, F.; Marlow, R.; Kanno, T.; Shinomiya, I.; Lombardi, S.; Buchupalli, B.; Graham, R.; Gazinska, P.; et al. Models of Breast Morphogenesis Based on Localization of Stem Cells in the Developing Mammary Lobule. Stem Cell Rep. 2015, 4, 699–711.
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In Vitro Propagation and Transcriptional Profiling of Human Mammary Stem/Progenitor Cells. Genes Dev. 2003, 17, 1253–1270.
- Slepicka, P.F.; Somasundara, A.V.H.; Dos Santos, C.O. The Molecular Basis of Mammary Gland Development and Epithelial Differentiation. Semin. Cell Dev. Biol. 2021, 114, 93–112.
- Dontu, G.; El-Ashry, D.; Wicha, M.S. Breast Cancer, Stem/Progenitor Cells and the Estrogen Receptor. Trends Endocrinol. Metab. 2004, 15, 193–197.
- Ercan, C.; van Diest, P.J.; Vooijs, M. Mammary Development and Breast Cancer: The Role of Stem Cells. Curr. Mol. Med. 2011, 11, 270–285.
- Economopoulou, P.; Kaklamani, V.G.; Siziopikou, K. The Role of Cancer Stem Cells in Breast Cancer Initiation and Progression: Potential Cancer Stem Cell-directed Therapies. Oncologist 2012, 17, 1394–1401.
- Charafe-Jauffret, E.; Ginestier, C.; Bertucci, F.; Cabaud, O.; Wicinski, J.; Finetti, P.; Josselin, E.; Adelaide, J.; Nguyen, T.T.; Monville, F.; et al. ALDH1-positive Cancer Stem Cells Predict Engraftment of Primary Breast Tumors and Are Governed by a Common Stem Cell Program. Cancer Res. 2013, 73, 7290–7300.
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927–3938.e6.
- Ginestier, C.; Monville, F.; Wicinski, J.; Cabaud, O.; Cervera, N.; Josselin, E.; Finetti, P.; Guille, A.; Larderet, G.; Viens, P.; et al. Mevalonate Metabolism Regulates Basal Breast Cancer Stem Cells and Is a Potential Therapeutic Target. Stem Cells 2012, 30, 1327–1337.
- Walsh, C.A.; Akrap, N.; Garre, E.; Magnusson, Y.; Harrison, H.; Andersson, D.; Jonasson, E.; Rafnsdottir, S.; Choudhry, H.; Buffa, F.; et al. The Mevalonate Precursor Enzyme HMGCS1 Is a Novel Marker and Key Mediator of Cancer Stem Cell Enrichment in Luminal and Basal Models of Breast Cancer. PLoS ONE 2020, 15, e0236187.
- Liu, S.; Clouthier, S.G.; Wicha, M.S. Role of microRNAs in the Regulation of Breast Cancer Stem Cells. J. Mammary Gland Biol. Neoplasia 2012, 17, 15–21.
- Monchusi, B.; Kaur, M. miRNAs as Modulators of Cholesterol in Breast Cancer Stem Cells: An Approach to Overcome Drug Resistance in Cancer. Curr. Drug Targets 2022, 23, 656–677.
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541.
- Sanders, M.E.; Schuyler, P.A.; Simpson, J.F.; Page, D.L.; Dupont, W.D. Continued Observation of the Natural History of Low-grade Ductal Carcinoma in situ Reaffirms Proclivity for Local Recurrence Even after More than 30 Years of Follow-Up. Mod. Pathol. 2015, 28, 662–669.
- Schuetz, C.S.; Bonin, M.; Clare, S.E.; Nieselt, K.; Sotlar, K.; Walter, M.; Fehm, T.; Solomayer, E.; Riess, O.; Wallwiener, D.; et al. Progression-specific Genes Identified by Expression Profiling of Matched Ductal Carcinomas in situ and Invasive Breast Tumors, Combining Laser Capture Microdissection and Oligonucleotide Microarray Analysis. Cancer Res. 2006, 66, 5278–5286.
- Tamimi, R.M.; Baer, H.J.; Marotti, J.; Galan, M.; Galaburda, L.; Fu, Y.; Deitz, A.C.; Connolly, J.L.; Schnitt, S.J.; Colditz, G.A.; et al. Comparison of Molecular Phenotypes of Ductal Carcinoma in situ and Invasive Breast Cancer. Breast Cancer Res. 2008, 10, R67.
- Wiechmann, L.; Kuerer, H.M. The Molecular Journey from Ductal Carcinoma in situ to Invasive Breast Cancer. Cancer 2008, 112, 2130–2142.
- Coradini, D. Interaction of de novo Cholesterol Biosynthesis and Hippo Signaling Pathway in Ductal Carcinoma in situ (DCIS)—Comparison with the Corresponding Normal Breast Epithelium. Transl. Breast. Cancer Res. 2023, 4, 26.
- Butt, S.; Butt, T.; Jirström, K.; Hartman, L.; Amini, R.M.; Zhou, W.; Wärnberg, F.; Borgquist, S. The Target for Statins, HMG-CoA Reductase, is Expressed in Ductal Carcinoma in situ and May Predict Patient Response to Radiotherapy. Ann. Surg. Oncol. 2014, 21, 2911–2919.
- Gill, S.; Stevenson, J.; Kristiana, I.; Brown, A.J. Cholesterol-dependent Degradation of Squalene Monooxygenase, a Control Point in Cholesterol Synthesis Beyond HMG-CoA Reductase. Cell Metab. 2011, 13, 260–273.
- Göbel, A.; Riffel, R.M.; Hofbauer, L.C.; Rachner, T.D. The Mevalonate Pathway in Breast Cancer Biology. Cancer Lett. 2022, 542, 215761.
- Kim, H.Y.; Bae, S.J.; Choi, J.W.; Han, S.; Bae, S.H.; Cheong, J.H.; Jang, H. Cholesterol Synthesis Is Important for Breast Cancer Cell Tumor Sphere Formation and Invasion. Biomedicines 2022, 10, 1908.
- Maja, M.; Mohammed, D.; Dumitru, A.C.; Verstraeten, S.; Lingurski, M.; Mingeot-Leclercq, M.P.; Alsteens, D.; Tyteca, D. Surface Cholesterol-Enriched Domains Specifically Promote Invasion of Breast Cancer Cell Lines by Controlling Invadopodia and Extracellular Matrix Degradation. Cell Mol. Life Sci. 2022, 79, 417.
- Tang, Q.; Liang, B.; Zhang, L.; Li, X.; Li, H.; Jing, W.; Jiang, Y.; Zhou, F.; Zhang, J.; Meng, Y.; et al. Enhanced Cholesterol Biosynthesis Promotes Breast Cancer Metastasis Via Modulating CCDC25 Expression and Neutrophil Extracellular Traps Formation. Sci. Rep. 2022, 12, 17350.
- Coradini, D.; Ambrogi, F.; Infante, G. Cholesterol de novo Biosynthesis in Paired Samples of Breast Cancer and Adjacent Histologically Normal Tissue: Association with Proliferation Index, Tumor Grade, and Recurrence-Free Survival. Arch. Breast Cancer 2023, 10, 187–199.
- Yoon, S.H.; Kim, H.S.; Kim, R.N.; Jung, S.Y.; Hong, B.S.; Kang, E.J.; Moon, H.G.; Noh, D.Y.; Han, W. NAD(P)-dependent Steroid Dehydrogenase-like is Involved in Breast Cancer Cell Growth and Metastasis. BMC Cancer 2020, 20, 375.
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene Expression Profiling in Breast Cancer: Understanding the Molecular Basis of Histologic Grade to Improve Prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272.
- Matikas, A.; Foukakis, T.; Swain, S.; Bergh, J. Avoiding over- and Undertreatment in Patients with Resected Node-Positive Breast Cancer with the Use of Gene Expression Signatures: Are We There Yet? Ann. Oncol. 2019, 30, 1044–1050.
More