1. The G Protein-Coupled Estrogen Receptor
In the late 1990s, a novel gene was identified in Burkitt’s lymphoma and breast cancer cells that encoded a heptahelical receptor that seemed to be associated with estrogen receptor alpha (ERα). This sequence showed homology to G protein-coupled receptors (GPCRs) and was named GPCR-Br
[1][2][1,2]. Further, this novel receptor was identified in animal models such as rats, where the homologous gene was detected in cells lacking ERα. These cells responded to 17β-estradiol (E2) stimulation via activating the mitogen-activated protein kinases (MAPK) pathway, and the receptor was named ER-X or GPR41
[3][4][3,4]. Nowadays, this receptor is known as G protein-coupled estrogen receptor 1 (GPER/GPR30) and has been identified as an alternative receptor for ERα that triggers cytoplasmic estrogen-signaling and promotes estrogen gene expression
[5][6][7][8][5,6,7,8].
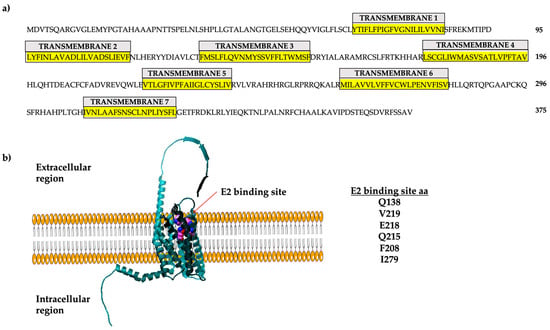
The human GPER gene is intronless, located on chromosome 7, region p22, and encodes a 375 amino acid protein
[9]. The GPER promoter region has estrogen response elements (EREs), which allow the binding of estrogen receptors to promote its transcription; consequently, E2 stimulates GPER expression
[10]. GPER is a plasma membrane receptor (
Figure 1) but can be internalized via a stimulus (e.g., E2) and localized in membranous organelles such as the endoplasmic reticulum and Golgi apparatus
[11][12][13][14][15][11,12,13,14,15]. GPER has seven transmembrane domains with extracellular N-terminal region. In contrast, the C-terminal region is cytoplasmic and interacts with hydrolases of nucleotide guanosine triphosphate (GTPases) or trimeric G-protein
[1][16][17][1,16,17]. GPER can be activated via E2 and inhibitors of ERα, such as tamoxifen (TMX), fulvestrant (ICI 182,780) (FVT), raloxifene (RLX), synthetic ligands (e.g., G1), aldosterone, and natural compounds such as kaempferol
[18][19][20][21][22][18,19,20,21,22]. These ligands activate GPER to trigger tissue-specific signaling. Some of these ligands are included in the therapeutic scheme against estrogen response cancer, which could explain the re-incidence or aggressiveness of cancer after treating patients.
Figure 1. GPER localization and amino acid sequence. (
a) The sequence of the 375 amino acids of the human protein is shown here. The seven transmembrane regions are indicated in yellow. (
b) Three-dimensional representation of GPER. The modeling was performed using Swiss Model with the template Q99527.1.A. Modified from
[9].
2. GPER/GPR30 Signaling in Normal Tissues
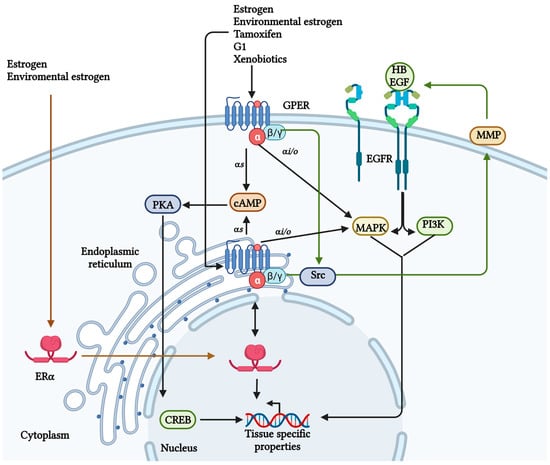
In humans, there are four different classes of G-protein alpha subunit (Gαs, Gαi/o, Gαq/11, and Gα12/13). It has been reported that GPER can be coupled to a Gα type s (Gαs) or Gα type i/o (Gαi/o); thereby, activating different routes of signaling and regulation
[6][23][24][25][6,23,24,25]. Once GPER is activated, the G-protein trimer is separated into two subunits, Gαs and Gβγ (
Figure 2). The Gαs subunit activates adenylate cyclase and cyclic adenosine monophosphate (cAMP) production to mobilize intracellular calcium (Ca
2+). Moreover, GPER decreases plasma membrane Ca
2+-ATPase (PMCA) activity, maintaining Ca
2+ in the cytoplasm and representing a secondary Ca
2+ regulation mechanism
[11][18][26][27][11,18,26,27]. This Ca
2+ regulation improves brain development, neuronal growth, and communication
[28], and also stimulates potassium (K
+) efflux, which promotes nitric oxide (NO) production in the endothelium, resulting in a vasodilator effect
[25][29][25,29].
On the other hand, GPER also seems to be associated with a Gαi/o because the pertussis toxin inhibits the GPER-mediated ERK ½ signaling
[25]. Also, the Gβγ subunit could transactivate the epidermal growth factor receptor (EGFR) via heparin-binding EGF-like growth factor (HB-EGF) activating the phosphatidylinositol 3-kinase (PI3K) and MAPK pathways (
Figure 2)
[30][31][30,31]. Furthermore, these mechanisms have been associated with the proliferation of non-tumorigenic breast cells MCF10A ex vivo
[32], dendritic spin density related to spatial learning and memory
[33][34][35][33,34,35], and glucose and lipid homeostasis
[36].
The binding affinity of E2 to GPER is 3–6 nM, whereas ERα’s is 0.1–1 nM. It is noteworthy that there has been no reported activation of GPER via E2 in epithelial and hippocampus models in vivo
[37]. However, other ligands, such as G1, activate the receptor
[38], showing the importance of using different study models. Nevertheless, as with other GPCRs which can be allosterically regulated, a GPER ligand-independent activity related to a domain involved in protein–protein interactions in the C-terminal, called PDZ motif, has been reported. This activation is associated with proliferation, migration, and invasion
[8][25][39][8,25,39]. These characteristics are related to neoplasia development, which suggests that GPER could be a cancer-relevant regulator.
Figure 2. GPER signaling. Different ligands can activate GPER; some are ERα ligands. Once GPER is activated, the G-protein trimer is separated into two subunits, the Gα is related to cAMP and calcium regulation, and the Gβγ can transactivate the EGFR and its signaling. However, a relationship exists between ERα and GPER, favoring the expression of genes related to cancer development
[11][30][40][11,30,40]. Created with BioRender.com (accessed on 25 August 2023).
3. GPER Expression in Cancer
GPER exhibits a tissue-specific activity which can be pro- or anti-tumoral, depending on the cancer type. The receptor level varies depending on its role; when it shows anti-tumoral activity (e.g., gastric, ovarian, and hepatic cancer), its expression is lower than in normal tissues. Research groups have reported that its activation or overexpression in these cancer types is related to a decrease in cancerous properties, supporting the anti-tumoral role of GPER in these cancer types
[31][41][42][31,41,42]. An example of this anti-tumoral activity is that GPER activation inhibits the growth of malignant cells through GPER/ERK signaling in hepatocarcinoma and xenograft models
[43]. Additionally, the low expression of this receptor in the hepatocarcinoma model has been related to promoter methylation and histone H3 deacetylation
[44]. Likewise, GPER anti-carcinogenic effects have been reported in prostate cells and xenografts involving G2 cell-cycle arrest
[45]. Moreover, the anti-proliferative activity of GPER has been observed in ovarian cancer
[46] and has been related to epigenetic regulation, such as the H3K4me3 mark
[41]. Furthermore, the GPER activation via tamoxifen inhibits proliferation, migration, and invasion in pancreatic cancer and regulates the tumorigenic environment, decreasing the anti-inflammatory macrophage phenotype M2
[47].
4. Epithelial–Mesenchymal Transition and Metastasis Development
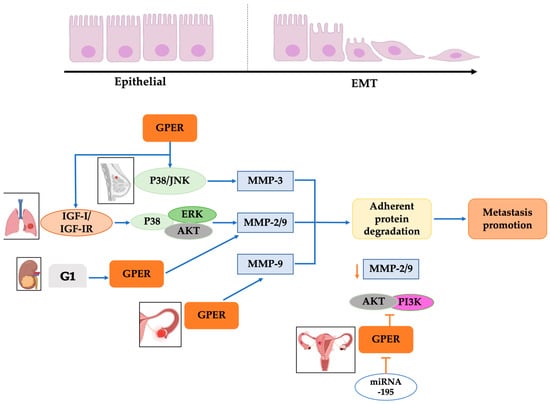
Metastasis is the dissemination of tumorigenic cells to other organs, allowing cancer cells to invade them and initiate a new tumor. This process requires phenotype plasticity to dedifferentiate mature cells back to progenitor states through the loss of linkage proteins and gain of mobilization phenotype, a phenomenon called epithelial–mesenchymal transition (EMT) (
Figure 3)
[48][62]. GPER has a relevant role in EMT because it promotes metalloprotease expression through the p38/JNK pathway, favoring the degradation of adherent proteins such as E-cadherin and acquiring a migratory phenotype in several types of cancer, such as breast, lung, colon, and ovarian
[31].
Figure 3. GPER’s relationship with EMT in cancer. Overexpression of GPER in breast, lung, kidney, and ovarian cancer cells generates a kinase activation pathway that stimulates MMP-3, MMP-2/9, and MMP-9 activation, which degrades the adhesive proteins, favoring the promotion of metastasis. Additionally, specific miRNAs, like miRNA195, block GPER activation in ovarian cancer cells; consequently, MMP-2/9 activation via AKT/PI3K is diminished, preventing the degradation of adhesive proteins and interrupting the promotion of metastasis. Modified from references
[31][49][50][31,49,52]. Created with BioRender.com (accessed on 25 August 2023).
5. Tumor Microenvironment Favors EMT through GPER
As aforementioned, EMT consists of re-programming expression patterns that stimuli of the environment can favor
[48][62]. The tumor microenvironment consists of normal cells, tumor cells, tumor stromal cells, including stromal fibroblasts, endothelial cells, immune cells such as macrophages and lymphocytes, and non-cellular components (
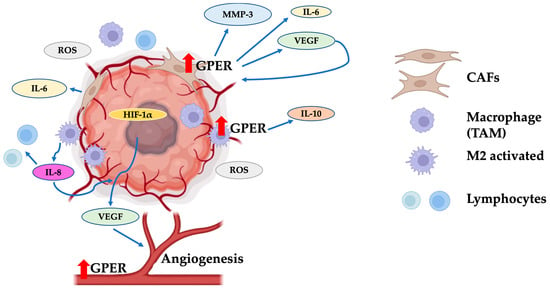
Figure 4). These elements are involved in a dynamic and bidirectional interaction between tumor cells and surroundings through secreted soluble molecules responsible for the horizontal information transfer between cellular/non-cellular communicating cells
[51][80].
The carcinoma-associated fibroblasts (CAFs) are normal fibroblasts activated via growth factors (e.g., transforming growth factor β (TGFβ), hepatocyte growth factor, fibroblast growth factor), and transcription factors (e.g., nuclear factor-kappa B (NF-κB) and heat shock factor-1 (HSF-1))
[52][81]. CAFs are a significant component of the tumor stroma and produce MMP-3 and cytokines that favor EMT
[53][82]. In addition, these cells express GPER, and its activation up-regulates IL-6, vascular endothelial growth factor (VEGF), and connective tissue growth factor (CTGF)
[27]. Also, the CAF secretome improves the expression of vimentin, ZEB1, and Snail in cancer cells, which are mesenchymal proteins essential for cancer progression
[22]. Consequently, the activation of GPER in CAFs stimulates the secretion of proteins that activate the cancer cells, promoting a re-programming of an epithelial expression pattern to a mesenchymal
[54][55][83,84]. Furthermore, CAFs promote TMX and FLV resistance in breast cancer and EMT in hepatocarcinoma via IL-6/IL-6R/STAT3 pathway
[56][57][58][85,86,87]. However, it has been reported that 3-methylcholanthrene activates the EGFR/ERK/c-Fos signaling in CAFs through an aryl hydrocarbon receptor (AhR)-GPER mechanism favoring the tumor growth via the up-regulation of cyclin D1 and CYP1B1
[59][56]. Also, GPER has a role in this resistance because the receptor levels are higher in drug-resistant cancer cells. However, when cells are exposed to receptor antagonists, cell sensibility is recovered
[14]. This resistance is related to an aggressive phenotype and mesenchymal protein expression, which suggests that GPER promotes drug resistance and cancer progression via EMT development.
Figure 4. The tumor microenvironment favors EMT through GPER. GPER overexpression in carcinoma-associated fibroblasts (CAFs) increases the production of MMP3, IL-6, and VEGF. These molecules improve tumor viability via inducing blood vessel formation. Additionally, tumor-associated macrophages (TAMs) increase IL-10 production through GPER activation, generating a protective microenvironment around the tumor mass. The expression of GPER on endothelium has been shown to influence the improvement of tumor angiogenesis. CAFs: carcinoma-associated fibroblasts; TAMs: tumor-associated macrophages; M2 activated: activated macrophages of type M2; ROS: reactive oxygen species; HIF-1α: hypoxia-inducible factor 1α; VEGF: vascular endothelial growth factor. Red arrows indicate un increase in the GPER levels. Modified from references
[53][60][61][62][75,77,82,88]. Created with BioRender.com (accessed on 25 August 2023).
6. Role of GPER in Angiogenesis
Angiogenesis involves creating new vessels that dispense nutrients and is a mechanism through which malignant cells can disseminate to other tissues. Endothelial cells are critical for tumor progression because vessel formation contributes to cancer dissemination and maintenance. Estrogens are related to vascular protection through receptors ERα, ERβ, and GPER
[63][98]. They are associated with protection in cardiovascular diseases, mainly in pre-menopausal females
[62][88], diminishing the inflammation and the NO production mediated in endothelial cells
[64][99]. The ERβ shows increased expression after vasculature injury in males and females and has been related to a protective effect
[65][100]. In addition, the estrogens are tightly linked to the angiogenesis process, stimulating the ERs
[66][101]. However, information about GPER’s protective activity in the endothelium is scarce. GPER mediates the activation of transduction pathways in different processes related to tumorigenesis, such as angiogenesis favoring tumor growth
[67][102] or stimulating endothelial cell proliferation
[68][103]. Some studies have shown that the activation of GPER via G1 inhibits the proliferation of endothelial cells, inhibiting DNA synthesis and arresting the cells in the S and G2 phases
[63][98]. Also, E2 and G1 activate the GPER/EGFR/ERK/c-fos signaling pathway and increase VEGF via up-regulation of HIF1α
[69][104], which is directly related to vessel formation. Also, E2 regulates endothelial cell tube formation in injury and has protective effects associated with GPER in endothelial cells
[66][101]. For this reason, GPER has been proposed as a possible therapeutic target to control the growth of some tumors, such as breast cancer.
7. Therapeutic Alternatives against Cancer Targeting GPER
Given the pro-tumorigenic role of GPER, it has been proposed as a therapeutic target. There are a variety of GPER antagonists, such as G15, G36, CIMBA, MIBE, PBX1, PBX2, C4PY, and CPT. The abovementioned drugs have been tested in breast cancer and CAF in vitro and in vivo, resulting in a GPER-dependent G1/E2-signaling inhibition
[70][71][105,106]. Additionally, chimeras made from an E3 ubiquitin ligase ligand called PROTACs degrade the target; in particular, the E2-PROTAC acts against GPER
[72][107]. However, GPER expression is simultaneous with ERα in some estrogen-responsiveness cancers; as a result, anti-estrogenic compounds must be reconsidered because drugs such as TMX and FVS act as a ligand for GPER
[20]. As mentioned, CAFs improve EMT; thus, novel therapies have been focused on them, but their use is related to severe secondary effects such as muscle loss and even death
[73][108]. On the other hand, triptolide is a drug reported as impairing the induction of EMT via CAFs in cancer cells. Also, there are novel therapies against signaling pathways related to the development of cancer properties, such as AG490, a drug against the JAK/STAT signaling involved in EMT that can be triggered via GPER activation
[74][109]. These findings suggest that GPER could be a promising therapeutic target that can act at different levels, regulating the cancer cell and its surrounding components.