Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ekaterina Georgieva and Version 2 by Jason Zhu.
SARS-CoV-2 infection, discovered and isolated in Wuhan City, Hubei Province, China, causes acute atypical respiratory symptoms and has led to profound changes in our lives. COVID-19 is characterized by a wide range of complications, which include pulmonary embolism, thromboembolism and arterial clot formation, arrhythmias, cardiomyopathy, multiorgan failure, and more. The disease has caused a worldwide pandemic, and despite various measures such as social distancing, various preventive strategies, and therapeutic approaches, and the creation of vaccines, the novel coronavirus infection (COVID-19) still hides many mysteries for the scientific community.
- COVID-19
- ROS
- RNS
- endothelial damage
- mitochondrial dysfunction
- Oxidative stress
- Thrombosis
- DIC
1. Endothelial Injury and Thrombosis
Damage to the epithelial lining of the airways through the release of cytokines and chemokines from immune cells is a hallmark of COVID-19. The underlying mechanisms of endothelial damage in COVID-19 are still being studied, but viral replication, the immune response, inflammatory mediators, and mitochondrial dysfunction are believed to be major contributors. Endothelial damage is a potential complication of COVID-19 and leads to impaired blood flow, increased risk of thrombosis, and ARDS. The severity of epithelial damage in COVID-19 can vary widely, with factors such as advanced age, chronic disease, and compromised immune status influencing the degree of epithelial involvement [1][106].
2. Oxidative Stress and Endothelial Dysfunction in Liver Sinusoidal Endothelial Cells
In the context of COVID-19, there have been reports and studies indicating that the virus can infect endothelial cells, which can lead to endothelial dysfunction and contribute to the complications seen in severe cases of liver disease [2][107]. Liver sinusoidal endothelial cells (LSECs) are a specialized type of endothelial cell found in the liver’s sinusoids. They possess unique fenestrations (small pores) in their cytoplasm, which allow for an efficient exchange of substances between the bloodstream and liver parenchymal cells (hepatocytes) and are considered the gatekeepers of liver homeostasis [3][108]. Many infections or inflammation can induce LSEC dysfunction, which can lead to disruptions in liver function and contribute to damage [4][109]. As an example, SARS-CoV-2 infection can trigger an inflammatory response and result in endothelial dysfunction, which may contribute to liver-related complications in COVID-19 patients [5][110]. In the case of COVID-19, there is evidence to suggest that the virus can infect endothelial cells, including LSECs. COVID-19 has been associated with an increased inflammatory response, which can lead to OS, and this can affect LSECs in several ways. High levels of RONS and redox imbalance can lead to damage to the fenestrae, the small pores in LSECs [6][111]. This damage can alter the structure and function of the fenestrae, reducing the efficiency of nutrient and oxygen exchange between the bloodstream and hepatocytes and causing an ineffective regulation of blood vessel tone modulation in the liver [7][112]. This can contribute to changes in blood pressure within the liver and impact overall liver function. Also, OS can trigger an inflammatory response within the liver, further exacerbating sinusoidal endothelial dysfunction and contributing to organ damage manifested as elevated liver enzymes (e.g., alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP)) and/or total bilirubin (TBIL)) [2][107].
3. Endothelial Damage of the Vascular Layer as a Result of Oxidative Stress in COVID-19
The endothelium is a layer of cells that lines the inner surface of blood vessels and plays an important role in regulating blood flow and preventing thrombotic events. Simultaneously, it actively participates in the production of various cytokines and adhesion molecules and is involved in key processes such as angiogenesis, coagulation, and regulation of vasomotor tone [8][113]. Endothelial dysfunction is characterized by increased permeability and edema formation, disruption of the balance between vasodilators and vasoconstrictors, and increased expression of adhesion molecules, as well as the release of ROS, etc. [9][114].
Chronic inflammation and OS are associated with the pathogenesis of various cardiovascular and cerebrovascular diseases such as atherosclerosis, hypertension, etc.; at the same time, they are considered to be major players in endothelial dysfunction, although the exact mechanisms are still not fully understood [10][11][115,116]. Endothelial dysfunction is thought to play a major role in the pathogenesis of COVID-19 and is associated with microangiopathy, pulmonary vascular changes, red blood cell microaggregates and platelet activation, and alveolar capillary microthrombi and endothelial damage [12][117]. In experimental models of hypertension, higher levels of ROS in blood vessels and oxidative damage to the vascular endothelium have been found [13][118]. Histopathological findings show that the pathology of COVID-19 includes not only hyperinflammation and cytokine storm, but also pronounced coagulopathy, impaired mitochondrial dynamics, and changes in vascular tone [14][119].
A spike in ROS levels initiates cellular damage and disrupts mitochondrial dynamics of platelet mitochondria and platelet function, which may induce hypercoagulation and thrombosis in COVID-19 [15][120]. On the other hand, the accumulation of dysfunctional mitochondria creates a prerequisite for the formation of free mitochondrial reactive oxygen species (mROS) and intermediate ROS products [16][121], an increase in nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) activity, and the inhibition of antioxidant signaling pathways [17][89]. Mitochondrial dysfunction is not limited to cellular mitochondria, but can also be observed in those free or membrane-enclosed platelets or vesicles [15][120]. The direct interaction of SARS-CoV-2 with endothelial cells increases the expression of inflammatory mediators such as IL-6, TNF-α, etc., which together with high levels of oxidants lead to endotheliitis, cell lysis, and apoptosis, disrupting the integrity on the vascular wall [18][122]. The endothelium performs a fine control of the vascular tone, participating in the maintenance of tissue hemostasis and vascular permeability. Endothelial NOS (eNOS) is mainly expressed in the cardiovascular system and is responsible for the production of nitric oxide (NO), which can interact with ROS and increase the formation of peroxynitrite. A disturbed NO/ROS balance causes increased vasoconstriction, oxidation, inflammation, thrombosis, and proliferation in the vascular wall and is one of the hallmarks of endothelial dysfunction [19][123].
SARS-CoV-2 viral pneumonia causes an overactivation of the immune response in lung tissue, which is almost always accompanied by OS, loss of redox control, and compromised mitochondrial function and dynamics, with subsequent dramatic endothelial damage [20][17] (Figure 1).
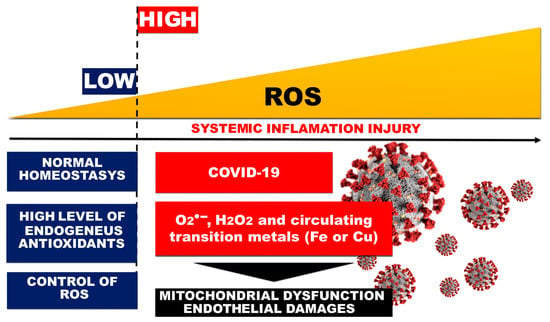
Figure 1.
Role of OS and systemic inflammation in COVID-19-related endothelial injury.
The SARS-CoV-2 virus can directly infect the endothelial cells of the vasculature, which potentiates cell damage and apoptosis [21][124]. This leads to a decrease in the antithrombotic activity of the vascular layer and an increase in factor VIII and von Willebrand factor (vWF). Elevated vWF reflects endothelial damage and indicates a high degree of platelet aggregation [22][125]. The close relationship between vWf and the processes of thrombus formation (thrombogenesis) or atherogenesis also suggests that high levels of vWf may be a useful indirect indicator of thrombosis [23][126]. Prevention of venous thrombosis is an important component of the complex and comprehensive treatment of COVID-19 and the post-COVID syndrome. Despite thromboprophylaxis in patients with coronavirus infection, the incidence of thromboembolic events is increasing [24][127].
4. DIC and Thrombosis in COVID-19 Cases
Thromboembolic events are associated with acute arterial ischemia due to obstruction of arterial vessels and occur despite the use of thromboprophylaxis and anticoagulant therapy [25][128]. Also, the overactivation of the coagulation system and inflammation in ARDS may increase the risk of pulmonary embolism, as the prevalence of extensive thrombosis and alveolar capillary microthrombi is significantly increased in COVID-19 [26][27][129,130]. Patients over 70 years of age are defined as particularly at risk for the development of arterial and venous thromboembolic events [28][131].
Disseminated intravascular coagulopathy (DIC) is a hemostatic disorder caused by various conditions such as sepsis, pancreatitis, trauma, and pregnancy [29][132], etc., as well as in patients with COVID-19 [30][31][133,134]. DIC occurs due to the abnormal activation of a cascade of reactions involved in blood clotting, leading to the activation of fibrinolytic mechanisms and deposition of fibrin in small vessels. Coagulation proteins and platelets may be depleted during the ongoing prothrombotic and fibrinolytic processes, leading to hemorrhage. Thus, in DIC, spontaneous bleeding and thrombosis can occur simultaneously, as an acute or chronic condition in different diseases [32][135].
The high mortality of patients with COVID-19 is due to various complications, including venous thromboembolism and disseminated intravascular coagulation (DIC) as a consequence of a systemic inflammatory response in the body [33][34][136,137]. Infection can lead to immunologically mediated vasculitis, expressed as an acute inflammatory reaction of the vascular layer, and causes thrombotic changes. Major risk factors include stasis, endothelial damage, and a hypercoagulable state known as Virchow’s Triad [35][138]. Milovanovic and colleagues consider endothelial damage, microvascular inflammation, and endotheliitis as the basis for severe COVID-19 infection and predictors of high mortality [36][139].
SARS-CoV-2 infection begins when viral spike proteins bind to ACE2, which initiates viral endocytosis into the endoplasmic reticulum. This increases the amount of Ang II and activates the enzyme NADPH oxidase. As a result, the production of superoxide radicals, hydrogen peroxide, and peroxynitrite is increased. Increased production of ROS and RNS affects the mitochondria, leading to the formation of mROS through oxidative and nitrosative damage to the mitochondrial electron transport chain. Mitochondrial ROS mediate various signaling pathways, increase the expression of inflammatory cytokines (IL-6, IL-1β, TNF-α, etc.), and increase the risk of thrombosis in patients with COVID-19 [37][140] (Figure 24).
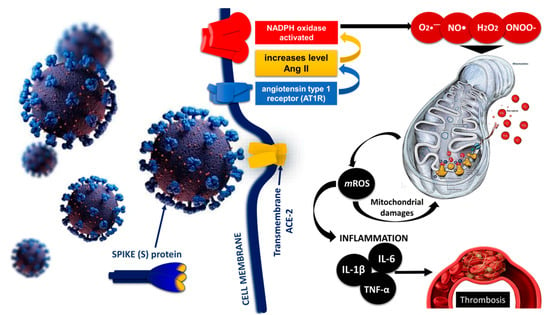
Figure 24. Role of mitochondrial dysfunction, mROS production, and inflammation in SARS-CoV-2 induced thrombosis [37][140] modified by Georgieva E et al. Hyperinflammation increases cytokine production, especially of IL-1, IL-6, and TNF-α, while ROS generated during cytokine storm can damage mitochondria and impair their function through oxidative damage to mtDNA.
Increased ROS formation leads to a disruption of the balance between antioxidants and oxidants, the activation of NADPH oxidase, and reduced NO bioavailability [13][118]. Low NO levels predispose the vasculature to a proinflammatory and prothrombotic state that can potentiate endothelial dysfunction [12][117]. As a result of the expression of proinflammatory cytokines and adhesion molecules, there is increased vascular permeability and hypertrophy of the vascular layer [38][141], loss of integrity of the epithelial–endothelial barrier, and passage of fluids and proteins to extravascular compartments, shortening of the half-life of proteins such as albumin [39][40][142,143]. A significant increase in IL-6 induces tissue factor (Factor III) expression in monocytes and macrophages, leading to thrombin generation [30][133]. In addition, inflammation contributes to thrombosis through endothelial damage and maintenance of a hypercoagulable state. Thrombi from extracorporeal membrane oxygenation contain more neutrophils than blood clots in other diseases [40][143]. Thrombosis and DIC are vascular events that are actively involved in the progression of COVID-19 and represent a coagulation/fibrinolytic abnormality manifested by macro- and microthrombotic events. The likelihood of the occurrence of COVID-19 thrombosis increases with increased markers of coagulation and fibrinolysis [41][42][144,145].