DNA damage repair lies at the core of all cells’ survival strategy, including the survival strategy of cancerous cells. Therefore, targeting such repair mechanisms forms the major goal of cancer therapeutics. The mechanism of DNA repair has been tousled with the discovery of multiple kinases. Studies on tousled-like kinases have brought significant clarity on the effectors of these kinases which stand to regulate double-strand break (DSB) repair.
- DNA damage and repair
- TLKs
- cancer
- cell cycle
1. Introduction
2. Role of TLK1 in DSB Repair
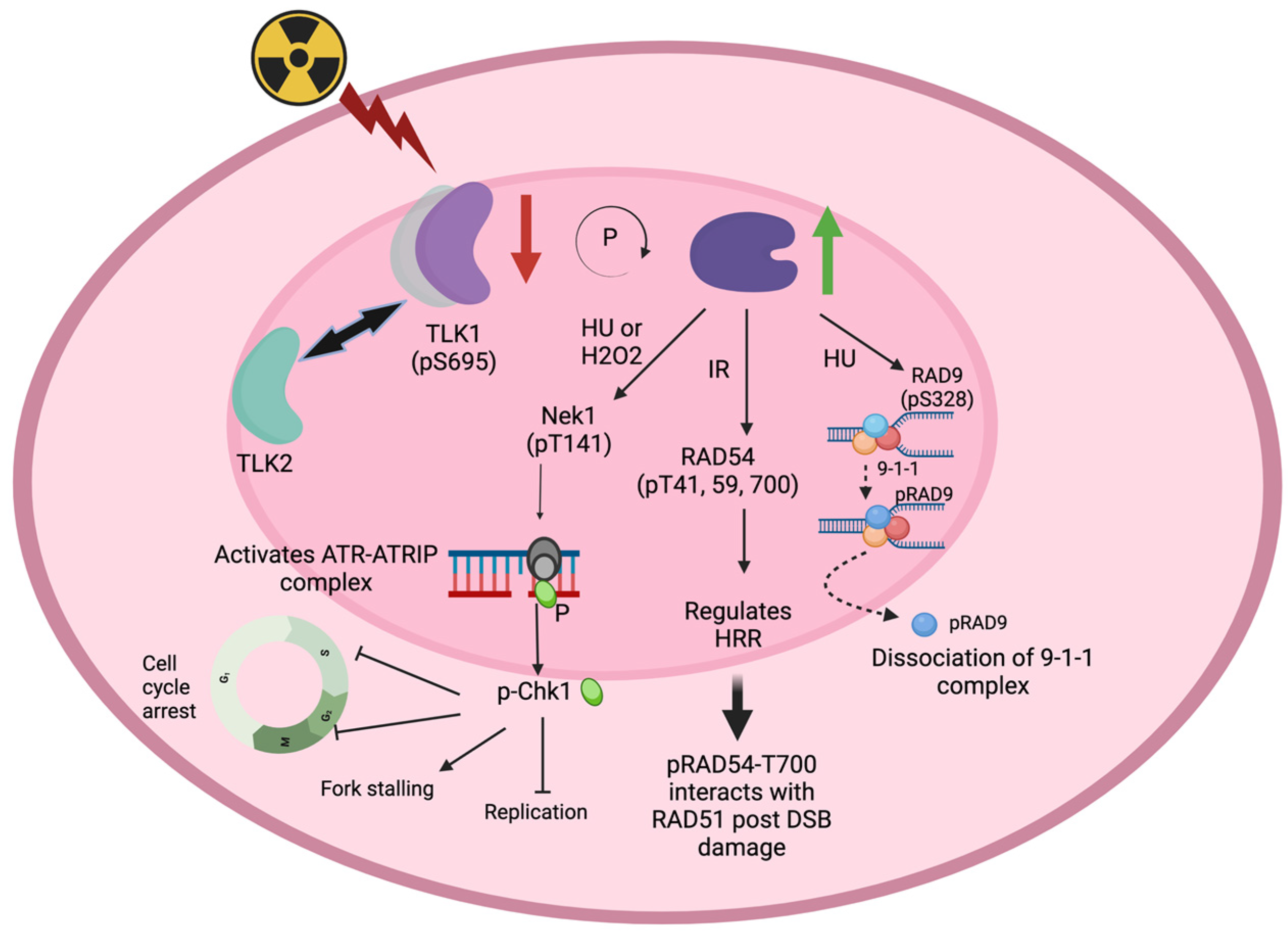
43. Role of TLK1 in Eukaryotic Recombination Repair
TLK1 activity is important for homologous recombination repair. Activation of TLK1 activity using gallic acid has been shown to increase HRR activity in cells [37]. TLK1 depletion by siRNA or shRNA methods across different cell lines has been shown to significantly decrease HRR efficiency in cells [4,35]. The TLK1 interactome reveals that RIF1 is a possible in vivo target [5,39,40]. RIF1 acts at the decision-making junction of NHEJ vs. HRR, downstream of 53BP1 [41,42]. It is known to turn on NHEJ while inhibiting 5′-end resections in HR. Asf1a/b (NTD, 1-154 a.a) has been found to interact with RIF1 (N-terminus, 967-1350 a.a), independent of its chaperone activity [43]. Although TLK1 interacts with both Asf1a/b and RIF1, it remains to be elucidated as to whether TLK1 can impinge on the NHEJ vs. HRR decision by regulating RIF1 and, further, whether it is dependent on TLK1 kinase activity or its chaperone function. In mammalian cells, RIF1 binds to aberrant telomeres in an ATM-53BP1-dependent manner when telomeres are unprotected and recognized as sites of DNA damage [44]. TLK1 depletion has been shown to increase telomeric sister chromatid exchange, thus indicating a state of hyper-recombination [45]. lizes to DNA damage foci in an RAD52-dependent manner [11,21]. In vitro, TLK1 phosphorylates RAD54B at T73. Interestingly, the localization of Rdh54 in kinetochores has been shown to be RAD52 independent. Since TLK1 has been shown to function in chromosome segregation in different organisms [35,46,47], it is speculated that TLK1 can regulate RAD54B functions in mitotic or meiotic events of chromosome dynamics. TLK1 preferentially localizes to the nucleolus even without DNA damage induction in HeLa cells [4].54. TLKs and DNA Damage and Checkpoint Functions
In addition to their functions in the repair of DSBs, TLKs are clearly involved in the repair process of other types of lesions such as UV-induced thymidine dimers (CPDs) [10] and cisplatin-induced ICLs [49]. While we still lack fundamental knowledge of their possible involvement in direct repair of these lesions, a possible explanation for their roles in the repair process, inferred through the effects observed in knock-down and overexpression studies, is, tentatively, their role in chromatin assembly and checkpoint functions. While several studies have described these activities, perhaps the most authoritative one was an unbiased siRNA-mediated screen study for kinases directly affecting potential regulators of recovery from DNA damage in U2OS cells, which positively identified TLK2 as modulating the DDR and G2 recovery. Furthermore, in TLK1 overexpression studies in normal mouse mammary cells, scholars directly observed in vivo that where MM3MG-TLK1B-overexpressing cells efficiently repaired almost all the CPDs in 12 h, control MM3MG cells showed very poor levels of CPD removal when assessed using the Southwestern blotting technique [10]. Another method (with intact cells) was also used to compare the repair abilities of the two cell lines. For this purpose, scholars assessed the repair of episomal vectors in cells over a time course following exposure to UV. MM3MG cells (that were stably transfected with the empty BK-shuttle vector) and the MM3-TLK1B cells (carrying the TLK1B insert in the same vector) were exposed to 5 J/m2 of UV radiation and allowed to recover for 0 to 12 h [10]. Episomes were extracted at various time points (0, 2, 4, 8, and 12 h) and were either mock-treated or digested with T4 endonuclease V enzyme, which specifically cuts DNA at CPD sites. Since the episomes were damaged on both strands at multiple sites, digestion with T4 endonuclease resulted in extensive cleavage of the recovered plasmid DNA, whereas, in unirradiated (or fully repaired) cells, T4 endonuclease left the plasmids intact.65. Role of TLK1 in Cancer
In different disease models such as prostate cancer (PCa) and glioblastoma (GBM) models, TLK1 depletion has been shown to elicit DNA damage response, and, therefore TLK1 forms a druggable target [51,52,53]. Human cancer data revealed frequent up-regulation of TLK genes and an association with poor patient outcomes in multiple types of cancer, and depletion of TLK activity led to increased replication stress and DNA damage in a panel of cancer cells [36]. GWAS analyses revealed that TLK1 mutations are rare in cancer, but its overexpression is frequently linked to poor prognosis [45,54]; this is particularly evident for patients with low Gleason scores (e.g., GS = 6—Ualcan.path.uab.edu/analysis page 2) who would otherwise be expected to fare better in terms of survival. Likewise, amplification of TLK2 has been reported as a frequent event in breast cancer [55], GBM [56], and neuroblastoma [57]. A previous work showed that DNA damage activates the TLK1 > NEK1 > YAP1 axis, which either further elevates the apoptotic pathway in PCa [20] or can lead to compensatory adaptation to genotoxins [17]. Broadly, it can be said that TLKs can impact cancer ontology and/or progression by regulating genome and epigenome stability [5], as well as potentially suppressing aspects of innate immune signaling [45].76. Targeting TLK1 for Cancer Treatment
After the seminal discovery that TLK1 has an important modulatory role in DDR and DNA repair, the quest turned rapidly into trying to identify specific TLK inhibitors to enhance the effectiveness of XRT or radiomimetic drugs. This led to the initial identification of certain phenothiazine (PTH) antipsychotics as surprisingly specific TLK inhibitors that, in fact, enhanced the killing of cancer cells, in vitro and in xenografts, when combined with IR or doxorubicin [38]. A newer PTH scaffold, called J54, that substantially lacks any anti-dopaminergic undesirable effect, showed very promising results in the regression of androgen-sensitive prostate cancer cells largely by passing the DDR and thereby enforcing entry into catastrophic mitotic progression, even resulting in substantial tumor regression in SCID xenografts [17]. Other studies on the structure/function of TLKs have revealed additional potential inhibitors [60]. Considering the availability of such drugs, of even greater importance now is the rebound effort to target the activity of TLK1 in RAD9 and RAD54, with the obvious suggestion that translesion (TSL) repair, single-strand gaps, and HRR can be simultaneously targeted. Various chemotherapeutic agents, therefore, fall under the umbrella of TLK inhibitors for their therapeutic potentiation. These include, for example: topoisomerase poisons, bleomycin, MMC, PARPis, and cisplatin, all of which ultimately lead to the formation of SSBs and DSBs. For some of these (e.g., doxorubicin and cisplatin), direct evidence of synthetic lethality in combination with TLK inhibitors has already been verified [38,61].