2. Adipose tissue Hyperplasia and Hypertrophy
Obesity is a metabolic state generated by adipose tissue expansion, which can occur through hyperplasia (proliferation and differentiation of adipose precursor cells) and hypertrophy (increase in adipocyte size), as demonstrated in
SchemeScheme 1 1 [13][13]. The number of adipocytes in a specific fat depot is primarily established early in life and tends to remain stable throughout adulthood
[14]. In contrast, adipocyte hypertrophy, caused by a constant surplus of energy, potential is remarkable and can reach a size increase of several hundred micrometers in diameter. Thus, adipose tissue expansion involves a complex interplay of factors including energy balance, genetics, and developmental processes
[15]. Adipose tissue expansion can be divided into two main phases: prenatal and postnatal. During the prenatal phase, adipose tissue undergoes dynamic changes characterized by both hyperplasia and hypertrophy processes
[16]. Upon gestational week 14, an early fat depot is starting to emerge from connective tissue. The primitive fat depots are composed of primary vessels and proliferating mesenchymal cells that subsequently will differentiate into preadipocytes
[17]. Adipocytes begin to appear by the 23rd gestation week, and by the 28th gestation week, discernible fat lobule structures are formed. At the end of pregnancy, fat depots contain and encompass diverse groups of adipocyte subpopulations, primarily distinguished by their fat content and, consequently, their size
[18][19][18,19]. The postnatal phase begins after birth and continues throughout life, with adipose tissue undergoing dynamic changes in response to energy balance and physiological needs by hyperplasia or hypertrophy
[14]. Adipose tissue hyperplasia and hypertrophy depend on and vary with age, with rapid hyperplasia and hypertrophy during early childhood (0–2 years) and adolescence (12–18 years), and with relative hyperplasia stabilization at adulthood, as was shown in longitudinal and cross-sectional studies by Knittle et al. and others
[20][21][20,21]. The rapid increase in both the hyperplasia and hypertrophy of adipose tissue occurs particularly in subcutaneous depots and is driven mainly by a growth hormone, insulin-like growth factor 1, sex steroids, and nutritional factors
[22][23][22,23]. Notably, childhood obesity is characterized by accelerated adipose tissue hyperplasia, resulting in the estimated doubling of the adipocyte number compared to normal-weight counterparts, which significantly elevate the risk for the development of obesity in later life as well
[23][24][23,24]. At normal weight states, the number of adipocytes peaks around puberty, and then stabilizes in adulthood. During adulthood, there is a gradual decline in adipose tissue hyperplasia potential with hypertrophy becoming predominant especially in visceral depots.
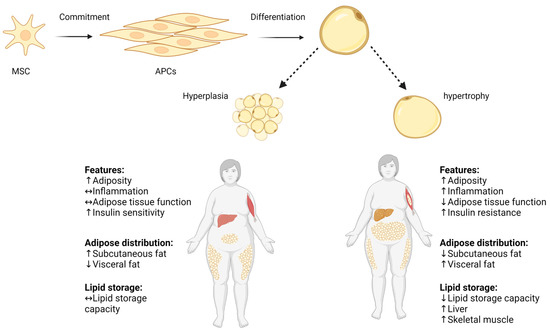
Scheme 1. Hyperplasia and hypertrophy in adipose tissue. Pluripotent MSC commit to differentiate into APC lineage located in the stromovascular fraction of adipose tissue, and APCs differentiate into mature adipocyte. Adipose tissue expands by increasing the volume of pre-existing adipocytes (adipose hypertrophy), and by generating new small adipocytes (hyperplasia). Increased adipocyte size correlates with impaired adipose tissue function, increased local inflammation, and decreased lipid storage capacity. ↑ increase, ↓ decrease, ↔ no change. Adapted from “Metabolically Unhealthy Obesity” and “Metabolically Healthy Obesity” by
BioRender.com (2022). Retrieved from
https://app.biorender.com/biorender-templates (accessed on 16 July 2023).
Earlier studies suggested that adulthood adipose tissue expansion is only through hypertrophic mechanisms
[25][26][25,26]. However, recent lineage-tracing models in rodents indicate that under an excess caloric intake, preadipocyte cells can also differentiate into new adipocytes, contributing to adipose tissue expansion via hyperplasia
[27][28][27,28]. Adulthood hyperplasia activation is possibly driven by the mature hypertrophic adipocytes’ limited oxygen diffusion capacity (which is at most 100 μm), and the neighboring cells’ mechanical stress
[29].
Adipose tissue growth, development, and distribution are also sex-dependently mediated by sex hormones such as estrogen and testosterone
[30][31][30,31]. For example, apart from estradiol fluctuation during the menstrual cycle, impacting appetite, caloric intake, and energy expenditure, it promotes subcutaneous fat accumulation, particularly in the gluteo-femoral region, resulting in the characteristic female pear-shaped body composition
[32][33][34][35][32,33,34,35]. Conversely, testosterone favors the deposition of visceral fat, leading to the typical male apple-shaped body composition
[36][37][36,37]. Moreover, subcutaneous adipose tissue usually has smaller more ‘plastic’ adipocytes, whereas the visceral adipose tissue is characterized by larger adipocytes encased in fibrotic tissues. Studies of rodents suggest that female mice present more adipose progenitor cells (APCs) in inguinal and gonadal depots compared to males, which respond to diet-induced obesity by elevating APCs’ hyperplasia (probably mediated by estrogen), while visceral fat expands with hypertrophy
[8][28][38][8,28,38]. The precise mechanism by which sex hormones determine adipocyte hyperplasia or hypertrophy is still not fully known
[30].
Hypoplasia is also a phenomenon related to adipose tissue
(although not the focus of the current review), characterized by the underdevelopment or reduced growth of adipose tissue. Hypoplasia results from various factors, including genetic mutations, hormonal imbalances, ageing, or inadequate nutrient availability
[39][40][39,40]. At the molecular level, adipose tissue hypoplasia can arise from differentiation, proliferation, and survival dysregulation through several key molecular regulators such as peroxisome-proliferator-activated receptor-gamma (PPARγ), CCAAT-Enhancer-Binding Proteins (C/EBPs), insulin-like growth factor 1 (IGF-1), and a fibroblast-growth-factor type (FGF). Additionally, inadequate nutrient availability, particularly essential fatty acids, can also limit adipose tissue growth
[41][42][43][44][41,42,43,44].
3. Adipocyte Hyperplasia
Adipose tissue comprises various cell types, including mature adipocytes, stromal cells, fibroblasts, macrophages, blood cells, endothelial cells, smooth muscle cells, mesenchymal stem cells (MSCs), and APCs. APCs, resembling fibroblasts, can differentiate into different preadipocyte lineages (e.g., beige and white adipocytes) in response to genetic and environmental factors, contributing to adipose tissue hyperplasia expansion
[45][46][47][48][45,46,47,48]. The pool of proliferating APCs is located in the stromovascular fraction of adipose tissue and is a depot and sex- and age-dependent
[31][40][45][49][50][31,40,45,49,50]. Ex vivo and in vivo human studies have identified APC types among CD31− and CD34+ stromal vascular fractions
[51][52][53][54][55][56][57][51,52,53,54,55,56,57]. Similarly, in a mice-specific sub-population characterized by cell surface immune markers was identified to possess proliferation and differentiation pro-adipogenic functional capabilities
[51][53][54][51,53,54]. Yet, the final commitment to the adipogenic linkage is not fully known. Adipocytes’ steady-state turnover rate has been under investigation with no consensus to date. In a constant nutritional condition, the suggested estimate is a stable 10% turnover rate yearly
[14]. This rate indicates that the maintenance of the adipocyte number normally involves a tightly regulated balance of adipogenesis and adipocyte death
[58]. However, shown by several elegant studies, at any life stage, states of over-nutrition lead to adipose tissue hyperplasia and, consequently, to an increased mass
[14][50][51][59][14,50,51,59]. Depot-dependent hyperplasia is a challenging research area due to technical issues; however, generally, it is accepted that a subcutaneous abdominal depot has a higher proliferative and differentiation capacity than a subcutaneous femoral depot and visceral adipose depot
[60][61][62][60,61,62]. WAT depot distribution plays a critical role and is a stronger predictor of metabolic health risks than overall obesity. The accumulation of fat in the visceral adipose depot and subcutaneous abdominal depot confers a higher risk of developing T2D and cardiovascular disease, while the accumulation of subcutaneous gluteal and subcutaneous femoral fat may be metabolically protective
[59][63][64][59,63,64].
Genetics also plays a role in an individual’s predisposition to adipose tissue hyperplasia, although
theour understanding on the subject is still evolving, and it is mostly investigated in animal models
[65][66][67][65,66,67]. Genetic factors contribute to the development and maintenance of adipose tissue by controlling processes such as cell proliferation, differentiation, and apoptosis. One of the key genetic factors playing a role in adipose tissue hyperplasia regulation is leptin. Leptin-recessive mutated mice (
Ob/
Ob) lack leptin, consequently exhibiting hyper-phagic, obesity, hyper-insulinemia, and hyper-glycemia
[68]. Furthermore,
Ob/
Ob mice present a larger number of adipose cells compared to the control, particularly being pronounced in the female mice
[65]. Findings in other genetically obese strains, such as New Zealand (NZO), yellow (
aAV), intermediate yellow (
aAfY), and (
db/
db), in states of a high- and low-fat diet show variation in obesity development, suggesting that diet-induced adipose tissue hyperplasia is strain-dependent, and indicating an interaction between genetics and nutrition
[69].
4. Adipocyte Hyperplasia in Obesity
During life, to replace mature adipocytes and under the condition of an excess energy flow, APC proliferate to generate new adipocytes
[70][71][70,71]. The proliferation of other stromal cells parallels this process to ensure a sufficient blood flow to supply oxygen and nutrients to the growing tissue
[72][73][72,73]. The number and size of adipocytes are critical factors in metabolic health. Many smaller adipocytes characterize a better healthy metabolic status, expressed by a better insulin sensitivity, a lower inflammation level, and less ectopic lipid accumulation
[74][75][76][74,75,76]. For example, in a prospective study of Pima Indians with either a normal, impaired, or diabetic glucose tolerance, Weyer et al. demonstrated that a smaller adipocyte cell size was positively correlated with an improved glucose tolerance
[76]. Enlarged adipocytes are typical of an obese state and correlate with the risk of metabolic syndrome (independent of BMI)
[77][78][79][77,78,79]. For example, the overexpression of insulin-responsive glucose transporter (GLUT4) in mice results in obesity and the expansion of body fat only with hyperplasia, accompanied by an improved glucose tolerance
[80]. Additionally, the collagen-IV-knock-out (KO) mice, characterized by unrestricted WAT hyperplasia under a positive energy balance due to the weakening of adipocytes’ extracellular scaffold, exhibit a significant weight gain yet an improved insulin sensitivity and inflammatory profile
[81]. WAT hyperplasia occurs in a depot-specific manner, as was shown in diet-induced obese mice models
[27][82][83][27,82,83]. WAT expansion in an intra-abdominal inguinal fat depot occurs almost exclusively through adipocyte hypertrophy
[28], possibly due to the microenvironmental conditions that suppress the APCs’ potential to undergo adipogenesis
[84]. In contrast, gonadal WAT can expand in both a hypertrophic and APC hyperplasic manner, triggered by nutrition and specific dietary lipids rather than the total caloric intake
[22][82][22,82]. Hyperplasia involves complex, sequential, and molecular mechanisms and can be divided into two distinguished processes:
The commitment step—the multipotent precursor mesenchymal stem cells (MSC) differentiate into APCs, which are committed to differentiate into preadipocytes’ lineage. The commitment step is regulated by several signaling pathways and cytokine, as well as epigenetic modifications such as DNA hypomethylation, insulin, glucocorticoids, transforming growth factor β (TGFβ) superfamily members, bone morphogenetic proteins (BMPs), and wingless (WNT) family members
[85][86][87][85,86,87]. The commitment stage is a complex process in which gene expression is precisely regulated and is extensively reviewed elsewhere
[88][89][90][88,89,90].
The differentiation step—preadipocytes undergo growth arrest, accumulate lipids, and form functional insulin-responsive mature adipocytes
[91]. The differentiation transcriptional regulation is a tightly regulated process, accompanied by transit and sequential expression at the level of different transcripts and proteins. This results in the progressive acquisition of morphological and biochemical characteristics of mature adipocytes
[92]. Preadipocytes respond to combined mitogenic and adipogenic signals necessary for the subsequent differentiation steps. In the early stage of adipocyte differentiation, the expression of C/EBPβ and C/EBPδ increases, which upregulates C/EBPα expression, further activating PPARγ, the master regulator of adipocytes’ differentiation. PPARγ binds to the retinoic acid X receptor (RXR) to form heterodimers that bind to the PPARγ response element (PPRE) and initiate the transcription of downstream genes, including C/EBPα (positive feedback), to obtain mature adipocytes’ phenotype with the ability to accumulate fat and secret adipokines, such as leptin and adiponectin
[93][94][93,94].
Adipose tissue hyperplasic expansion is a process regulated by hormones through endocrine, paracrine, autocrine, and neural systems
[95][96][97][95,96,97]. Centrally, hormones and cytokines regulate satiety/hunger, metabolic, and activity states in a complex net of interactions. Major central players include glucagon-like peptide-1 (GLP-1), neuropeptide Y, leptin, ghrelin, and Cholecystokinin (CCK)
[98]. Peripherally, molecular regulators of the adipose cell number include insulin, PPARγ ligands, retinoids, corticosteroids, and tumor necrosis factor-alpha (TNFα)
[92][99][100][92,99,100]. Additionally, adipose tissue is innervated by sympathetic neurons, where APCs’ proliferation is highly responsive to β-adrenergic signaling
[101].
States of dysregulation or unbalanced regulation of the complex cascade manifesting adipose tissue development can lead to accelerated adipocyte differentiation and hyperplasia
[92][102][92,102]. For example, adipocytes are among the most insulin-responsive cell types, thus critically contributing to whole-body insulin sensitivity and energy homeostasis
[103][104][103,104]. Furthermore, insulin is an obligatory hormone in preadipocyte differentiation to mature adipocytes in both in vivo and in vitro models
[105][106][105,106]. Obesity induces an insulin resistance state, which is characterized by an adipocyte expansion blockage, possible death, and hyperplasia contributing to obesity co-morbidities, including T2D
[107][108][107,108]. Growth factors such as insulin-like growth factor 1 (IGF-1) and fibroblast growth factor type 1 (FGF1) are crucial regulators of cell proliferation, survival, and differentiation and participate in the regulation of hyperplasia in adipose tissue
[109][110][111][109,110,111]. Alteration in growth hormone levels can lead to the dysregulation of the activation of downstream signaling pathways such as the phosphatidylinositol 3-kinase (PI3K)/AKT and the mitogen-activated protein kinase (MAPK) pathways, which are known to stimulate cell growth and proliferation
[112]. Several studies have shown negative correlations between BMI and absolute IGF-1 levels, while others reported increased circulating IGF-1 in obesity and, specifically, abdominal obesity due to elevated portal insulin levels
[113][114][115][116][117][113,114,115,116,117].