Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Elisa Pinto and Version 2 by Rita Xu.
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy. The hypervascular nature of the majority of HCCs and the peculiar vascular derangement occurring during liver carcinogenesis underscore the importance of angiogenesis in the development and progression of these tumors. Indeed, several angiogenic molecular pathways have been identified as deregulated in HCC. The hypervascular nature and the peculiar vascularization of HCC, as well as deregulated angiogenic pathways, represent major therapeutic targets.
- hepatocellular carcinoma
- angiogenesis
- tyrosine kinase inhibitors
1. Introduction
Hepatocellular carcinoma (HCC) represents the sixth most commonly diagnosed cancer and the third leading cause of cancer-related death globally [1]. Approximately half of HCC patients are diagnosed at advanced tumor stages, precluding potentially curative treatments such as surgical resection or liver transplantation [2]. As a consequence, the prognosis of patients with HCC is very poor with 5-year survival of 20% [3].
Angiogenesis, one of the fundamental hallmarks of cancer [4], plays a pivotal role in the development and progression of HCC, which is typically a hypervascular tumor [5][6][5,6]. Since, the growth of liver tumor requires the formation of new blood vessels, HCC displays intense neoangiogenic activity during its development. Moreover, a peculiar vascular derangement occurs during liver carcinogenesis, since the tumor tends to be almost entirely fed by arterial inflow, unlike the surrounding parenchyma that receives the majority of blood supply through the portal system [7]. However, in liver tumors, newly formed blood vessels display marked vascular abnormalities, which may further activate angiogenic pathways, leading to a vicious cycle. It has been demonstrated that the overactivation of angiogenesis in HCC is associated with worse prognosis. A transcriptomic signature of five genes involved in the angiogenetic process (ANGPT2, NETO2, ESM1, NR4A1, and DLL4) was found to accurately identify rapidly growing tumors and was associated with shorter survival [8]. In addition, several studies suggest that overexpression of vascular endothelial growth factor (VEGF) and its transcription factor hypoxia-inducible factor (HIF)-1α, the two key mediators of angiogenesis, is a negative prognostic factor, particularly in patients treated with surgery and systemic therapies [9][10][11][12][13][14][15][16][17][18][19][20][9,10,11,12,13,14,15,16,17,18,19,20].
The very important role of angiogenesis in the development and progression of HCC provides a strong rationale for antiangiogenic strategies as therapy. Angiogenesis has always been considered an important therapeutic target in these patients. Intra-arterial locoregional treatments (IATs) (i.e., transarterial embolization (TAE) and transarterial chemoembolization (TACE)) are commonly applied treatments for HCC worldwide [21]. Their activity is completely or in part reliant on the embolization of tumor feeding arteries with the aim of achieving tumor ischemic necrosis. Considering systemic therapies, over the last decades, multiple antiangiogenic therapies have been developed. In fact, most currently approved treatments for advanced HCC in the first- and second-line settings target angiogenic pathways [22]. More recently, the combination of an immune checkpoint inhibitor (ICI) anti-programmed death ligand 1 (PD-L1) (atezolizumab) and a monoclonal antibody targeting VEGF (bevacizumab) demonstrated a clear survival benefit over sorafenib [23]. Considering that previous trials with anti-PD1 immune checkpoint inhibitors alone (nivolumab and pembrolizumab) failed to show efficacy in first- and second-line treatment [24][25][24,25], these results seem to further confirm the importance of angiogenic pathways in the progression of HCC.
2. Angiogenesis in Hepatocellular Carcinoma
Normal liver receives approximately 26% of cardiac output (~1.1 mL O2/g/min) and consumes approximately 20% of the total O2 used by the body at rest (~0.06 mL O2/g/min) [26]. About 75% of the hepatic blood supply is received by the portal vein, while the rest is received by the hepatic artery. Lobules are the fundamental unit of normal liver: they are segregated by interlobular connective tissue and contain “cords” of hepatic parenchymal cells (hepatocytes), separated by vascular sinusoids. Sinusoidal endothelium is fenestrated and lacks a basement membrane, therefore permitting blood plasma to surround hepatocytes through the space of Disse. Other cells involved in liver physiology are hepatic stellate cells, also known as pericytes, and Kupffer cells, which are resident liver macrophages. Stellate cells are closely linked to sinusoids in the space of Disse and play a crucial role in liver fibrosis after liver damage. In the hepatic sinusoids, arterial (from the hepatic artery) and venous (from the portal vein) blood mix together, and after being “filtered” by hepatocytes, this blood flows out of the lobule through the central hepatic vein. In liver tumors, newly formed blood vessels display marked vascular abnormalities [27][28][27,28], leading to hypovascular areas and severe hypoxia and/or necrosis and causing further stimulation of angiogenesis. Although HCC is a highly angiogenic cancer, it seems to be characterized by hypoxia [5], which has been associated with HCC growth, progression and resistance to therapies [29]. Nevertheless, while some characteristics of HCC (hypervascularity, areas of necrosis and primary resistance to therapy) suggest the presence of severe hypoxia, direct evidence of hypoxia in human HCC is missing [26]. In fact, pO2 in human HCC have not yet been accurately measured directly, and thus, the relevance of hypoxia in determining hypervascularity and arterialization of HCC is still unproved [26]. Moreover, it is important to keep in mind that the activation of angiogenic pathways (HIF target genes) can be achieved by various hypoxia-independent mechanisms [30]. The destabilization of the microvasculature, leading to vascular hyperpermeability, remodeling of the extracellular matrix and endothelial cell activation, is a fundamental step for the initiation of angiogenesis [5]. Activated endothelial cells form new blood vessels by proliferating, migrating and undergoing cord formation. Subsequently, recruited and activated pericytes stabilize the newly formed blood vessels [31][32][33][31,32,33]. During physiological angiogenesis, the release of antiangiogenic molecules balances the expression of proangiogenic factors [34]. By contrast, as shown in Figure 1, tumor-induced angiogenesis results from an imbalance between proangiogenic factors (VEGF-A, -B, -C and -D, angiopoietins, fibroblast growth factor (FGF), hepatocyte growth factor, endoglin (CD105), platelet-derived growth factor (PDGF), and others) and anti-angiogenic molecules (angiostatin, thrombospondin-1, endostatin, and others) [22][22]. In the following paragraphs, the roles of the main molecules involved in angiogenesis are reviewed briefly.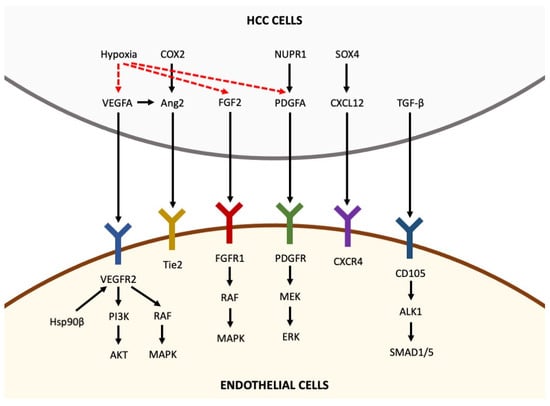
Figure 1. Pro-angiogenic factors inducing angiogenesis in hepatocellular carcinoma. Pro-angiogenic factors, including VEGFA, Ang2, FGF2, PDGFA, CXCL12 and TGF-β, are secreted by HCC cells and bind to their receptors expressed in endothelial cells, thus activating intracellular pathways that promote angiogenesis. Hypoxia is able to upregulate the expression of VEGFA, FGF2 and PDGFA in HCC cells (the arrows indicate the sequentiality of molecular pathways).