Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Robert T. Mallet and Version 2 by Sirius Huang.
High-altitude illnesses (HAIs) result from acute exposure to high altitude/hypoxia. Numerous molecular mechanisms effect appropriate acclimatization to hypobaric and/or normobaric hypoxia and curtail the development of HAIs.
- altitude
- hypoxia
- acclimatization
- oxidative stress
- redox homeostasis
- mitochondria
- genes
1. Introduction
Acute exposure to high altitude imposes hypobaric hypoxia, raising the risk of high-altitude illnesses (HAIs) in inadequately acclimatized individuals [1][2][3][1,2,3]. Such illnesses include acute mountain sickness (AMS), the most frequently observed but usually benign and self-limited form of HAI, and the rare but life-threatening high-altitude cerebral edema (HACE) and high-altitude pulmonary edema (HAPE) [1][2][1,2]. Although hypobaria per se may contribute to HAI symptoms [4][5][4,5], the central pathogenetic factor is hypoxia [1][6][7][8][1,6,7,8]. Appropriate acclimatization to hypoxia, whether hypobaric or normobaric, can minimize or even prevent those illnesses [2][9][10][2,9,10].
Although systemic physiological responses to acute hypoxia and systemic physiological changes occurring during acclimatization are well established after decades of extensive research [11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29], the molecular mechanisms driving these physiological adjustments are not fully understood [30][31][32][33][34][35][30,31,32,33,34,35]. The discovery and characterization of hypoxia-inducible factors (HIFs), key transcription factors regulating gene expression when cellular oxygen availability declines, and other molecular pathways mediating hypoxia responses [36] have paved the way to a better understanding of molecular processes underlying acclimatization to high altitude/hypoxia [37]. For their contributions to deciphering the molecular mechanisms of oxygen sensing in cells and tissues, William G. Kaelin Jr., Sir Peter J. Ratcliffe, and Gregg L. Semenza were awarded the 2019 Nobel Prize in Physiology or Medicine [38]. Nevertheless, our understanding of the complex cell- and tissue-specific signaling and signal integration leading to high-altitude acclimatization on a systemic level is still in its infancy, in particular when considering differences among individuals and additional environmental factors, such as temperature, humidity or radiation.
2. Systemic Physiological Responses to Acute High-Altitude Exposure and Acclimatization
The most important physiological responses to acute high altitude/hypoxia exposure comprise hyperventilation triggered by the hypoxic ventilatory response (HVR) [39], hemoconcentration due to enhanced diuresis, and an increase in heart rate and cardiac output as a result of sympathetic activation in hypoxia/at altitude [6][40][6,40]. In contrast to peripheral vasodilation, acute hypoxia exposure causes pulmonary vasoconstriction (HPV) and elevated pulmonary arterial pressure [17].
Ventilatory acclimatization to hypoxia (VAH), the progressive increase in ventilation during acclimatization, supports recovery of the initially reduced alveolar (PAO2) and arterial (PaO2) oxygen partial pressures and arterial oxygen saturation (SaO2) [12]. Increased ventilation lowers alveolar (PACO2) and arterial (PaCO2) partial pressures of carbon dioxide, producing respiratory alkalosis and compensatory renal bicarbonate excretion [41]. Paralleling the increased PaO2 and SaO2, both hemoconcentration and increased cardiac output (at rest and during exercise) help to maintain tissue oxygen delivery in the hypoxic high-altitude environment [42]. However, the acclimatization process, whether occurring at high altitude or in artificial environments (hypobaric or normobaric chambers, tents or face masks with reduced inspired oxygen), encompasses—in addition to ventilatory acclimatization—a broad spectrum of physiological changes, e.g., cardiovascular, cerebrovascular, metabolic, hematological, neurophysiological, and hormonal adjustments [2][10][43][44][45][2,10,43,44,45]. Figure 1 depicts the time courses of selected physiological responses to moderate or high altitude/hypoxia.
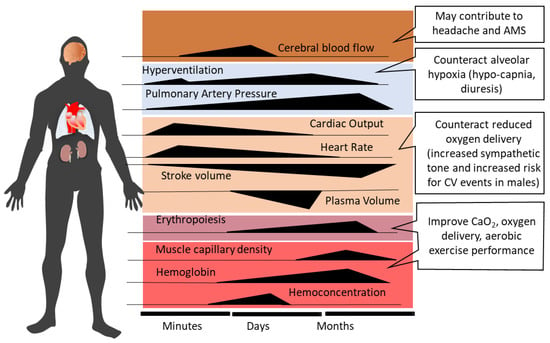
Figure 1. Time-dependent changes and related consequences of physiological responses during acclimatization to high altitude (modified from Burtscher et al., 2022 [10]). AMS, acute mountain sickness; CV, cardiovascular; CaO2, arterial oxygen content.
Alveolar minute ventilation progressively increases over the first 8–10 days at altitude and then plateaus [12][46][12,46]. Sympathetic activation at the onset of acute altitude/hypoxia exposure acutely increases heart rate and systemic blood pressure [47][48][47,48]. Over the following 10 days at altitude, heart rate tends to decrease [48][49][48,49], while elevated systemic blood pressure persists [47]. Increased PAO2 with acclimatization may produce some dampening of HPV [50][51][50,51]. As the initial hemoconcentration subsides, HIF-1 related upregulation of erythropoietin (EPO) sustains the increases in hemoglobin (Hb) concentration and arterial oxygen content (CaO2 = Hb x SaO2) after the first 1–2 weeks of high-altitude exposure [52]. The increase in muscle capillarity due to vascular endothelial growth factor (VEGF) up-regulation [53] represents another HIF-1-initiated response to altitude/hypoxia exposure.
3. High Altitude Illnesses: Epidemiology and Pathophysiology
This section provides a brief overview of the heterogeneous pathophysiologic processes involved in the development of HAIs, which are largely determined by the diverse responses and susceptibility to hypoxia-associated injury among individuals. A comprehensive understanding of the pathophysiology of HAI is essential for the development of new treatment options, for example, to facilitate successful acclimatization by targeting adaptive or maladaptive signaling pathways and molecular processes.
3.1. Acute Mountain Sickness and High-Altitude Cerebral Edema
Acute mountain sickness (AMS) frequently develops in high-altitude visitors and typically follows a benign disease course, with symptoms that often resolve over a few days of acclimatization [2][54][2,54]. AMS incidence steeply increases with an ascent in altitude or hypoxia intensity (from about 7% at 2200 m to over 50% at 4559 m) in susceptible individuals not acclimatized to high altitude/hypoxia [55][56][55,56]. The incidences of severe AMS and HACE are about 24% and 1%, respectively, at 4000 m altitude [57].
Unacclimatized individuals typically develop AMS symptoms within the first 6 to 12 h of acute high-altitude exposure [1][2][1,2]. While headache represents the cardinal symptom, subjects most susceptible to AMS typically develop nausea first [58]. In persons suffering from HACE, altered mental status and ataxia are the most prevalent symptoms and are often accompanied by headache, anorexia, nausea, vomiting, and retinal hemorrhages [59][60][59,60]. AMS development in unacclimatized persons may be ascribable to acute cellular and/or systemic responses that are delayed and/or insufficient to maintain oxygen delivery to tissues at high altitudes [2][12][39][2,12,39].
Diagnosis of AMS is usually based on the Lake Louise Scoring system (LLS) or less frequently on the abridged (11-item) version (ESQ-C) of the 67-item Environmental Symptoms Questionnaire (ESQ-III) [2]. The original LLS [61] is a self-assessment questionnaire, rating the severity (no discomfort = 0; mild symptoms = 1; moderate symptoms = 2; severe symptoms = 3) of five main criteria, i.e., headache, nausea, dizziness, fatigue, and difficulty sleeping, but has recently been revised by deleting the “difficulty sleeping” criterion [62].
Increased intracranial pressure, brain swelling and edema formation may constitute AMS pathophysiology and explain the symptoms [63][64][65][63,64,65]. Sequential magnetic resonance imaging (MRI) scans during a 22-h exposure to normobaric hypoxia (FiO2: 0.12) demonstrated total brain parenchymal expansion, but only the extent of white matter edema (indicating vasogenic edema) correlated with AMS severity [63]. These authors suggested that veno-compression of the small and deep cerebral veins likely contributes to elevated intracranial pressure and brain swelling [63]. Major AMS symptoms, i.e., headache, may result from activation and sensitization of the trigemino-vascular system via mechanical (e.g., elevated cranial and/or intravascular pressure) and/or biochemical (e.g., reactive oxygen species (ROS), nitric oxide (NO), prostaglandins, inflammatory molecules) factors [19][66][67][68][19,66,67,68].
Why and when severe AMS progresses to HACE remains unclear. Although AMS and HACE are often considered manifestations on a continuum of cerebral HAIs [69], with mild AMS likely progressing to severe AMS and, in rare cases, even to HACE [69], whether HACE really is a severe form of AMS [1] or rather an independent entity remains to be established. The occurrence of white matter edema in HACE indicates dysfunction or disruption of the blood-brain barrier (BBB), which may be caused by ROS-related membrane destabilization and inflammation and/or local HIF and VEGF activation [70].
Pharmacological treatment options for AMS primarily aim to improve oxygen delivery (e.g., hyperoxic breathing or augmenting hyperventilation by the use of acetazolamide), reduce AMS symptoms (e.g., headache) and/or prevent cerebral edema formation by administration of dexamethasone and/or nonsteroidal anti-inflammatory drugs [1][2][71][1,2,71]. However, prevention of AMS in particular, and HAIs in general, through appropriate acclimatization strategies is the most effective countermeasure.
3.2. High-Altitude Pulmonary Edema
Both HACE and HAPE represent life-threatening diseases with ~50% mortality when untreated [71]. Like AMS and HACE, the incidence of HAPE primarily depends on the rate of ascent, the absolute altitude attained and the individual’s susceptibility to HAPE. For instance, in mountaineers with unknown HAPE history who ascended over 4 days to an altitude of 4500 m, the HAPE incidence was 0.2%, but the incidence increased to 6% when ascending in only 1–2 days [71], underscoring the importance of acclimatization to prevent HAPE.
HAPE is a non-cardiogenic pulmonary edema caused by pronounced hypoxic pulmonary vasoconstriction and related elevations of pulmonary-artery and capillary pressures, resulting in a noninflammatory and hemorrhagic alveolar capillary leak [72]. Genetic predisposition has been implicated as a cause of the remarkably robust pulmonary vascular responses to hypoxia in some individuals, which may be attributed to insufficient formation and bioavailability of NO likely associated with high ROS levels in hypoxia/at altitude [73][74][73,74].
While in high-altitude pulmonary hypertension (HAPH), inflammatory pathways may importantly contribute to the proliferation of pulmonary artery smooth muscle cells and pulmonary hypertension [75], this may not be the case for HAPE. Although inflammation in HAPE could contribute to increased alveolar-capillary permeability, studies in humans rather indicate that inflammation constitutes a secondary response to the pulmonary edema and/or disruption of the alveolar-capillary barrier, not a primary factor in HAPE pathogenesis [72].
Besides rapid descent and oxygen supplementation, administration of the calcium channel blocker nifedipine has been shown to be effective for HAPE prophylaxis and treatment [17][76][17,76], but others have questioned nifedipine’s effectiveness [77].
It must be mentioned that apart from hypoxia, several other environmental and/or behavioral factors could contribute to the heightened risk of serious illnesses at high altitudes [78]. Although a comprehensive examination of these factors and their interactions with hypoxia are beyond the scope of this text, the potential contributions of hypothermia and dehydration to HAIs merit discussion. For example, exposure to cold at high altitudes may predispose mountaineers to dehydration due to elevated cold-diuresis and poor access to fluids [79]. These authors identified an association between the level of dehydration and the risk of AMS [79]. Moreover, exposure to cold and altitude may increase the risk of thrombosis and myocardial infarction [80]. Heavy exercise at altitude exacerbates dehydration, raising the risk of rhabdomyolysis and acute liver injury [81], and intensifying arterial hypoxemia, thereby triggering or accelerating the development of AMS [82]. Furthermore, both severe low ambient temperature and intense physical activity are also implicated as predisposing factors for HAPE development, primarily due to the increase in pulmonary artery pressure [83][84].[83,84]