Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Richard Mbi Beteck | + 3184 word(s) | 3184 | 2021-05-19 05:22:09 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Beteck, R. Novel Antituberculosis Agents. Encyclopedia. Available online: https://encyclopedia.pub/entry/9927 (accessed on 28 June 2026).
Beteck R. Novel Antituberculosis Agents. Encyclopedia. Available at: https://encyclopedia.pub/entry/9927. Accessed June 28, 2026.
Beteck, Richard. "Novel Antituberculosis Agents" Encyclopedia, https://encyclopedia.pub/entry/9927 (accessed June 28, 2026).
Beteck, R. (2021, May 21). Novel Antituberculosis Agents. In Encyclopedia. https://encyclopedia.pub/entry/9927
Beteck, Richard. "Novel Antituberculosis Agents." Encyclopedia. Web. 21 May, 2021.
Copy Citation
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a curable airborne disease currently treated using a drug regimen consisting of four drugs.
tuberculosis
drug development
pharmacokinetics
1. Introduction
Tuberculosis (TB) is a contagious bacterial infection—one of the top ten causes of death, especially in young adults worldwide [1]. TB is the leading cause of mortality from a single infectious agent, ranking above human immunodeficiency virus (HIV) and malaria [2]. Mycobacterium tuberculosis (Mtb), a bacterium discovered by Dr. Robert Koch in 1882, is the causative agent of TB, and it is a member of the Mycobacterium tuberculosis complex (MTBC). MTBC is composed of several subspecies responsible for TB in mammals [3]; while all these subspecies were isolated, only five of them are known to commonly cause TB in humans [4]. The primary organs of Mtb’s aerobic infection are the lungs, resulting in pulmonary TB. From the lungs, the infection can spread to other organs (brain, bone marrow, and spine), leading to extrapulmonary TB [1]. The common mode of TB transmission is through droplet nuclei from an infected person to others, which are generated during coughing, sneezing, or speaking [5].
Although TB affects both HIV-positive and HIV-negative people, the number of deaths estimated in HIV-negative people outweighs the former. There were an estimated 1.3 million TB-related deaths among HIV-negative people and 300,000 deaths among HIV-positive people in 2018 [2]. This amounts to nearly 2 million TB-related deaths every year, a number that has been relatively stable in recent years [2]. Co-infection with HIV infection greatly increases the chances of an individual developing active TB following exposure, and also immunosuppression related to uncontrolled HIV infection leads to reactivation of latent TB [6]. Additionally, Smoking, diabetes, and malnutrition increase susceptibility to active TB; silicosis, organ transplant, tumor necrosis factor-alpha inhibitors are other risk factors for reactivation of latent TB [7].
In addition to the above-mentioned risk factors, antimicrobial resistance (AMR), mainly the multidrug-resistant Mtb (MDR-Mtb), extensively drug-resistant Mtb (XDR-Mtb), and totally drug-resistant Mtb strains (TDR-Mtb) are also implicated in the increasing TB prevalence to an extend [8]. The problem of MDR-TB and XDR-TB across the world has become very alarming as a direct consequence of mistakes and adherence to the prescribed chemotherapy, availability of anti-TB drugs, surveillance of the patients, and incorrect administration of anti-TB drugs [9]. Other factors contributing to the rise in TB cases include increased immigration from TB endemic countries to countries with low TB prevalence [10] as well as the growing level of homelessness coupled with drug abuse [11].
Presently, treatment of drug-sensitive (DS) TB is divided into a two-month intensive phase of treatment comprising of rifampicin, isoniazid, pyrazinamide, and ethambutol (first-line drugs) followed by a four-month continuation phase consisting of rifampicin and isoniazid [12]. Failure of first-line drugs is attributed to several reasons, including non-compliance of patients (affecting clinical and a microbiological cure) [13] and side effects of prescribed drugs [14]. This failure often leads to the emergence of MDR-TB, defined as resistance to isoniazid and rifampicin, the two most potent first-line drugs in the TB treatment regimen [15]. Consequently, the need arises to treat MDR-TB with relatively expensive and more toxic second-line drugs. Moreover, treatment of MDR-TB often requires a longer treatment period (minimum of up to 18 months) [4,7]. The current second-line drugs are grouped into three groups: A, B, and C, according to their efficacy profiles [16]. Group A drugs include the fluoroquinolones (Levofloxacin or moxifloxacin), bedaquiline (BDQ), and linezolid; group B includes clofazimine, and cycloserine or terizidone; group C contains ethambutol, delamanid, pyrazinamide, imipenem-cilastatin or meropenem, amikacin or streptomycin, ethionamide or prothionamide, and p-aminosalicylic acid [17,18].
Resistance of Mtb to any fluoroquinolone and to at least one of the injectable drugs (capreomycin, kanamycin, or amikacin), in addition to isoniazid and rifampicin resistance, is termed XDR-TB [15]. Notably, co-infection of HIV and XDR-TB strains are increasingly associated with poor treatment outcomes in HIV-seropositive persons [19].
The compliance and toxicity issues associated with first-line drugs, the emergence and worldwide spread of multidrug-resistant Mtb, the high failure rate of drug candidates in the development pipeline, and the general propensity of bacteria to develop resistance against newly approved drugs make paramount the continuous search of novel compounds with antitubercular activity. To promote the discovery of new antitubercular agents, we herein compiled the different classes of drug candidates in lead optimization, pre-clinical, clinical development, and recently approved drugs, as well as their putative targets (see Scheme 1 below). We search articles from the internet using keywords, such as new TB agents, TB drug pipeline, TB drug candidates, and pharmacokinetic of TB drug candidates. We also consulted sites such as the Working Group on New TB drugs, TB Alliance. New or repurposed compounds under investigation from 2010 onward were also selected.
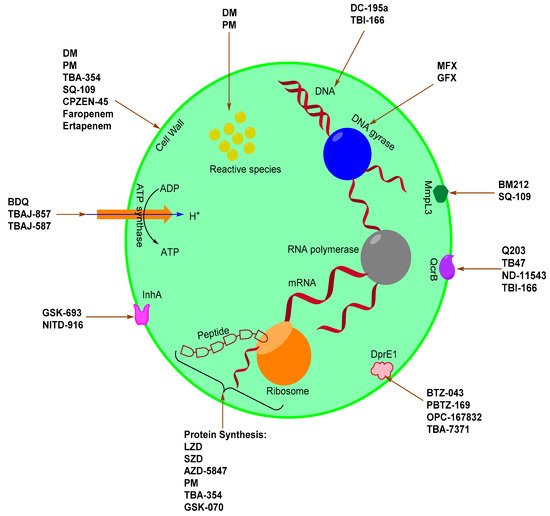
Scheme 1. TB drugs and drug candidates and their associated targets.
2. Novel Anti-Mtb Agents
Mtb has a well-developed cell wall which provides an extraordinary lipid barrier. This barrier facilitates the manifestation of either acquired or intrinsic resistance to antibiotics [20]. It is imperative to understand the mode of action of current drugs as well as the mechanisms of bacterial resistance in the pursuit to discover or develop new anti-TB drugs or regimens aimed at combating difficult-to-treat bacterial strains (MDR and XDR) [20,21]. The mode of action for existing and emerging drugs against Mtb include inhibition of the cell wall, cell wall acids, and peptidoglycan (WecA) synthesis, DNA gyrase and topoisomerases, DNA replication, protein synthesis, ATP synthase, Lipid synthesis, DprE1, InhA, QcrB, LeuRS, MmpL3 protein, and L,D-transpeptidase.
2.1. InhA Inhibitors
Enoyl-acyl carrier protein reductase (InhA) is a key enzyme in the type II fatty acid biosynthesis pathway (FASII) in Mtb. InhA was first identified as a clinical target based on the therapeutic potential of isoniazid in TB treatment [235,236]. InhA catalyzes the NADH-dependent reduction in long-chain trans-2-enoyl-acyl carrier proteins (ACPs) [235]. Isoniazid, an essential component of the TB drug regimens, targets the InhA of Mtb, and it is a prodrug activated by KatG—a mycobacterial catalase-peroxidase enzyme [237,238]. The dependency of isoniazid on KatG activation is the main clinical weakness (particularly drug resistance) associated with its application due to mutation occurrences in katG [239]. Thus, identifying inhibitors that directly bind to InhA without the requirement for activation by KatG (direct InhA inhibitors) may represent a valid strategy to overcome isoniazid resistance in TB treatment [240]. GSK-693 and NITD-916 (Figure 11) are novel direct InhA inhibitors of Mtb currently studied as potential substitutes for isoniazid in current TB treatment regimens.
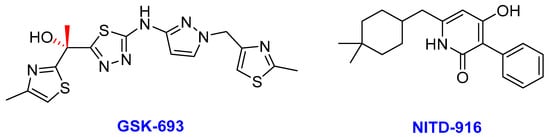
Figure 11. The chemical structures of GSK-693 and NITD-916; novel direct Mtb InhA inhibitors.
- (i)
-
GSK-693
Lead optimization studies by GlaxoSmithKline (GSK) led to the identification of the thiadiazole chiral compound GSK-693 as a novel, selective, and promising lead compound with attractive antitubercular properties that overcome isoniazid resistance. GSK and other research units have carried out high-throughput screening against InhA using a collection of GSK compounds and identified the thiadiazole series to be the most promising compound class [241]. Despite the interesting in vitro antitubercular profile obtained from the initial SAR studies, hits were affected by a number of compound development requirements such as physicochemical properties, drug metabolism, and pharmacokinetics (DMPK) profiles [241].
Further tests confirmed that GSK-693 does not require KatG activation and retain the antitubercular activity against DS, MDR, and XDR clinical isolates [241]. Like other InhA direct inhibitors, thiadiazoles bind to the enzyme–NADH complex, but they do not establish any direct interaction with active site residue Tyr158 [242]. Unlike the isoniazid-NADH adduct, GSK-693 does not cause the flipping of the Phe149 side chain, and there is no interaction with the isonicotinic acid-binding pocket [241]. In addition, compounds inhibiting InhA without requiring activation by KatG could be active under anaerobic conditions where catalase-mediated activation is suppressed by the lack of oxygen.
GSK-693 displayed equal potency (MICs of 0.2 µg/mL) against the Mtb H37Rv inside and outside of macrophages, along with a good CYP 3A4 inhibition profile [241]. The compound demonstrates decreased lipophilicity, enhanced solubility, and reduced metabolic liabilities coupled with simultaneous good oral bioavailability at different doses in acute and chronic mouse models [165]. Lastly, early safety assessments done across different enzymatic assays indicated that GSK-693 did not show any sign of cytotoxicity and no hERG potassium channel inhibition [243], suggesting a low risk for general toxicity and cardiotoxicity observed in isoniazid.
- (ii)
-
NITD-916
More efforts to overcome isoniazid resistance in Mtb led to the discovery of a new class of direct InhA inhibitors called 4-hydroxy-2-pyridones, identified using phenotypic high-throughput screening [241,244]. The lead compound of the 4-hydroxy-2-pyridones series known as NITD-916 exhibits good activity against isoniazid-resistant MDR-TB clinical isolates and displays good efficacy in vivo in acute and established mouse TB infection models [95]. To inhibit InhA, NITD-916 forms a ternary complex with InhA-NADH, which blocks access to the enoyl substrate-binding site with a consequential reduction in the biosynthesis of mycolic acids and eventually cell death [245,246]. Based on the co-crystal structure, the 4-hydroxy-2-pyridones (including NITD-916) interact with the InhA–NADH complex through hydrogen bonding, pi-stacking, and hydrophobic interactions [247]. However, this mechanism of InhA inhibition was recently challenged by Flint et al. (2020) using killing kinetic studies to suggest that inhibition of InhA is bactericidal against nutrient-starved non-replicating Mtb, regardless of the binding mechanism [248]. This corroborates earlier findings that isoniazid reduces CFU in starved bacteria [249,250].
NITD-916 displays superior MIC against Mtb H37Rv over isoniazid (0.08 µg/mL vs. 0.8 µg/mL respectively) [251]; and also proves to have a lower frequency of resistance (1 × 10−8) than isoniazid (1 × 10−5) in vitro [95]. The difference in frequency of resistance is because isoniazid is a prodrug, and mutations in the KatG enzyme occur at a high frequency [251]. To this effect, NITD-916 overcomes a key liability associated with the application of isoniazid in current TB treatment regimens [251]. Thus, portraying potential as a novel TB therapeutic agent that is useful in guiding the development of improved hydroxy-pyridines and other direct InhA inhibitors.
2.2. β-Lactams
The β-lactams are the most populous class of antibiotics and are the mainstay of most antibacterial drug treatment regimens; however, their efficacy against Mtb has always been limited [72]. Nonetheless, β-lactams generally offer a well-defined safety profile than other second-line alternatives [252]. Therefore, β-lactams: faropenem and ertapenem (Figure 12) briefly discussed below are being studied in phase II clinical trials [37].
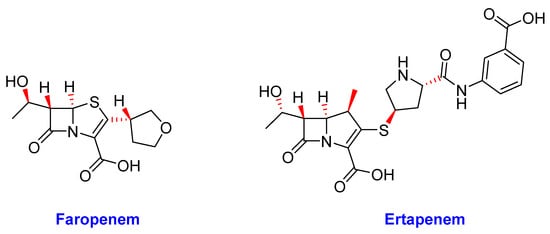
Figure 12. The chemical structures of faropenem and ertapenem.
- (i)
-
Faropenem
Faropenem is an orally bioavailable (72–84%) penem antibiotic that is more resistant to hydrolysis by β-lactamases than cephalosporins and carbapenems [253]. It is structurally similar to the carbapenems; however, this penem ring system is slightly less strained and consequently has improved chemical stability [254]. Additionally, it has been modified to a prodrug ester (faropenem medoxomil), which permits oral administration, both of which are desirable advantages for treating MDR-TB [72]. The drug is already approved for the treatment of respiratory infections in humans, but it also displayed promising bactericidal activity in both active and non-replicating Mtb cells comparable to meropenem [72,253].
Faropenem demonstrates potent biochemical activity independent of clavulanate (β-lactamase inhibitor) and inactivate Mtb L,D-transpeptidase enzymes more efficiently than meropenem [253]. Thus, the drug inhibits the L,D-transpeptidases, which perform the last cross-linking step of peptidoglycan synthesis. The MIC of faropenem against Mtb is 1.3 µg/mL with or without clavulanate, as opposed to an 8-fold increase in MIC for meropenem and many folds for other β-lactams tested in the absence of clavulanate [253]. Whilst using the dehydropeptidase inhibitor, probenecid; Dhar and colleagues demonstrated that Mtb-infected mice had a small but significant reduction in lung burden (7.7 and 7.5 log10 CFU/mouse at the beginning and end of treatment, respectively) after 9 days of treatment with a combination of faropenem/clavulanate/probenecid [253,255].
In summary, faropenem’s stability, oral bioavailability, superior biochemical potency against its molecular target, and proven efficacy in the treatment of infections caused by nontuberculous mycobacteria all justify its potential in the chemotherapy of MDR-TB and XDR-TB [256].
- (ii)
-
Ertapenem
The second β-lactam agent to have shown promising clinical results and favorable pharmacokinetic properties in the treatment of Mtb is ertapenem, a member of the carbapenem class of antibiotics [151]. Amongst all β-lactams, the carbapenems are the most suitable for treating Mtb as they potently target the high molecular weight penicillin-binding proteins and also inactivate the unusual L,D-transpeptidases that form the 3→3 crosslinks found in the unique Mtb cell wall [253,257]. Carbapenems inhibit the peptidase domain of penicillin-binding proteins, leading to autolysis and peptidoglycan weakening of the cell wall [258]. The development of carbapenems for TB treatment has raised considerable interest recently because the presence of clavulanic acid establishes uniform activity against XDR-Mtb and kills both growing and dormant forms of the bacilli [258]. The carbapenems are less favorable substrates for the Mtb β-lactamase BlaC due to rapid acylation and slow deacylation [72]. As such, unlike other β-lactams, they are not rapidly hydrolyzed by BLaC and thus maintain their activity against Mtb [259]. Although encouraging results have recently been obtained using meropenem for the treatment of XDR-TB in humans, the compound is unstable and has poor bioavailability [253]. As a result, frequent intravenous administrations of the drug are required, making its general clinical application limited [165].
Following reconstitution and dilution, Ertapenem degradation is temperature-dependent. Thus, the proposed in-use shelf life for ertapenem is 6 h at room temperature, 24 h at 2–8 °C [151]. Since Mtb has a doubling time of at least 24 h under the best of circumstances [260], microbial killing and inhibition of growth by the most effective of antibiotics, especially by β-lactams, are slow (requiring several days) because the drugs depend on cell wall turnover [151]. Consequently, drugs such as ertapenem, already appearing unstable at 37 °C [259], are likely to be degraded before killing or inhibiting slow-growing bacteria, especially semi-dormant Mtb. However, drug susceptibility tests show that ertapenem is likely to have a good sterilizing effect in TB [151]. This might be attributed to its rapid inactivation of L,D-transpeptidases (which is more rapid than imipenem and meropenem), as well as due to its reversible protein binding nature [257]. To reach maximal bactericidal activity for Gram-positive, Gram-negative, and anaerobic bacterial infections, ertapenem is given intravenously at a dose of 1000 mg once daily [261]. Ertapenem has a long plasma half-life of 4 h, compared to other carbapenems, enabling once-daily dosing, which is an advantage for MDR-TB treatment [262,263]. The AUC0–24 of ertapenem is 544.9 h·mg/L with a Cmax of 127.5 mg/L in MDR-TB patients [259].
Generally, ertapenem treatment is well tolerated during MDR-TB treatment [259,264]. There are no drug-drug interactions, and the drug is not metabolized by cytochrome P450, nor is it a substrate for p-glycoprotein [265,266]. Renal excretion is the main route of elimination for ertapenem. Therefore, the pharmacokinetics of the drug are altered to a significant extent clinically in patients with severe renal impairment [267]. In such cases, a dose reduction to 500 mg once a day is recommended [266].
2.3. Oxoborates
Recently, boron-containing compounds known as oxaboroles have been shown to inhibit LeuRS (Leucyl-tRNA synthetase) by the oxaborole tRNA-trapping mechanism [268]. The aminoacyl-tRNA synthetases are a family of essential enzymes that are required for protein synthesis in all cells [269]. The boron atom is an integral part of Mtb LeuRS inhibitors, as it forms a bidentate covalent adduct with the terminal adenosine nucleotide Ade76 of tRNALeu [269]. Consequently, this covalent adduct traps the 3′ end of the tRNALeu in the editing site forming a nonproductive complex, blocking leucylation and bacterial protein synthesis [268].
- (i)
-
GSK-3036656 (GSK-070)
Another product of potential interest discovered by the GSK group is GSK-3036656, alternatively known as GSK-070 (Figure 13) is currently in a phase II clinical trial for the treatment of TB [270]. GSK-070 is a novel 3-aminomethyl-4-chloro-benzoxaborole with inhibitory activity against the Mtb enzyme LeuRS [271]. Thus, GSK-070 binds to the catalytic site of LeuRS that is responsible for the hydrolysis of incorrectly ligated aminoacylated tRNAs [179]. Notably, the binding is necessitated by the presence of the amino group of the (S)-aminomethyl side chain at C-3 through hydrogen-bonding interactions [271]. Whereas the halogen substituents (Cl or Br) improves the Mtb’s LeuRS and antitubercular activity against Mtb H37Rv as well as selectivity against other bacteria [272]. GSK-070 exhibited promising activity against laboratory strains of Mtb as well as against selected DS-TB, MDR-TB, and XDR-TB clinical isolates [271].
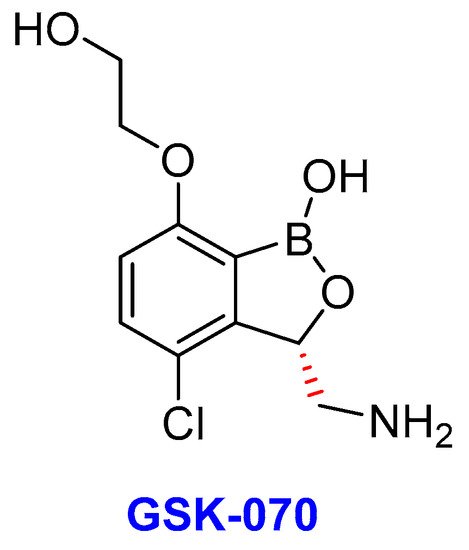
Figure 13. The chemical structure of GSK-070, an oxoborate drug in TB clinical trials.
The efficacy of GSK-070 was assessed against replicating and nonreplicating/slow-replicating mycobacteria in mice models of acute and chronic Mtb H37Rv lung infections [273,274]. In the acute model, a maximum difference of 3.6 log10 CFU was obtained in the lungs compared to untreated mice for 8 days; whereas the chronic cohort produced a drop of 2.1 log10 CFU after 2 months of daily oral treatment at maximum doses of 1.1 and 1.3 mg/kg, respectively [273]. A recent phase 1 clinical study evaluating the safety, tolerability, and pharmacokinetics of single and repeat oral doses of GSK-070 met all primary endpoints and showed no serious adverse events [179,275]. The study determined potent inhibition of Mtb LeuRS (IC50 = 0.20 µM) and in vitro antitubercular activity (Mtb H37Rv MIC = 0.08 µg/mL) [272]. GSK-070 showed a dose-proportional increase following single-dose administration after dosing for 14 days. Additionally, the drug showed 2–3 fold increase in Cmax and AUC accumulations with repeated administration after the dosing period that was unaltered in the presence of food [271]. Furthermore, GSK-070 is a low-hepatic-clearance compound with high solubility and moderate passive permeability and is not a p-glycoprotein substrate; hence it undergoes rapid absorption and exhibited very high oral bioavailability in the nonclinical species (mouse, rat, and dog) [271]. Finally, in vitro data suggest that GSK-070 has a low plasma protein binding in the nonclinical species (mice, rats, and dogs, and humans) [271]. The main route of elimination for GSK-070 is in the urine.
Table 1 below summarizes the mutation event(s) associated with resistance against novel anti-TB drugs and drug candidates.
Table 1. Mutant genes and Mutations causing Mtb resistance to novel anti-TB drugs under development.
| Drug | Mutant Gene | Mutation(s) | References |
|---|---|---|---|
| MXF | gyrA | D94G, D94N, and D94Y | Nosova et al. 2013; Groll et al. 2009 |
| GFX | gyrA gyrB |
A90V, A94G, A94T, A94A, A94H and A89A Δ678, Δ679, and A533T |
Nosova et al. 2013; Groll et al. 2009 |
| BDQ | atpE rv0678 |
A63P, A63V, D28A, D28V, D28P, D28N, D28G, R124stop, L40F, T91P, and E21stop G66E, M1A, W42R, S53L, S53P, S63R, and S63G |
Huitric et al. 2010; Segala et al. 2012; Zimenkov et al. 2017; Andries et al. 2014; Pang et al. 2017; Xu et al. 2018; Zhang et al. 2015 |
| LZD | rplC rrl |
C154R G2299T, G2814T, G2270T, and G2746A |
McNeil et al. 2017; Zhang et al. 2016; Pang 2017; Balasubramanian et al. 2014, Zhang et al. 2014; McNeil et al. 2017 |
| DM | ddn fgd1 |
L107P and 59–101 (deletion) T960C |
Schena et al. 2016, Fujiwara et al. 2017 |
| PM | ddn fgdi fbiC |
V616, Y89H, and Y133D R212Q A2158A and C1114T |
Haver et al. 2015 |
| SQ-109 | mmPL3 | A700T, L567P, Q40R, and T2055375C | Tahlan et al. 2012 |
| BTZ-043 | dprE1 | C387G, C387S, C387A, C387T, and C387N | Foo et al. 2016 |
| PBTZ-169 | dprE1 | C387G, C387S, C387A, C387T, and C387N | Foo et al. 2016; Chen et al. 2020 |
| OPC-167832 | dprE1 (rv3790) mmpS5-mmpL5 (rv0678) |
C387G, C387S, C387A, C387T, C387N, and V388T G248C, A364S,T314H, and 84–85InsIS6110 |
Hariguchi et al. 2020; Milano et al 2009 |
| TBA-7371 | dprE1 | Y314H | Gawad and Bonde 2018 |
| Q203 | qcrB | T313A | |
| NITD-916 | inhA fabG1InhA |
S19W, I21M, I21V, F41L, F47L, S94A, M103T, D148E, M161L, R195G, I202F, G205S, G205A, G205R, A206E, G212D, G214P, I215S, L269R, Δ210, T162M, and R49H C-15T |
McNeil 2017 |
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
947
Revision:
1 time
(View History)
Update Date:
21 May 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No