 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Irene Bulli | + 1916 word(s) | 1916 | 2021-05-19 04:59:04 | | | |
| 2 | Catherine Yang | Meta information modification | 1916 | 2021-05-21 03:54:39 | | |
Video Upload Options
Ischemic stroke is a leading cause of death and disability worldwide. The only pharmacological treatment available to date for cerebral ischemia is tissue plasminogen activator (t-PA) and the search for successful therapeutic strategies still remains a major challenge. The loss of cerebral blood flow leads to reduced oxygen supply and a subsequent switch to the glycolytic pathway, which leads to tissue acidification. Carbonic anhydrase (CA, EC 4.2.1.1) is the enzyme responsible for converting carbon dioxide into a protons and bicarbonate, thus contributing to pH regulation and metabolism, with many CA isoforms present in the brain. Recently, numerous studies have shed light on several classes of carbonic anhydrase inhibitor (CAI) as possible new pharmacological agents for the management of brain ischemia.
1. Introduction
2. Carbonic Anhydrase Inhibitors (CAIs) as Possible Therapeutics in the Central Nervous System Pathologies
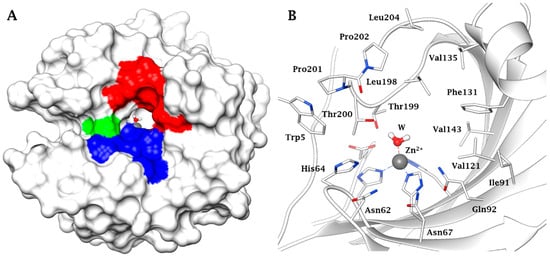
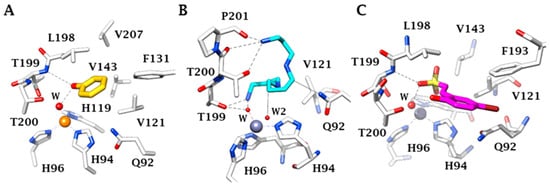
3. Protective Mechanisms of CAIs in Cerebral Ischemia
References
- Dirnagl, U.; Iadecola, C.; Moskowitz, M. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397.
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; Van Der Worp, B.H.; Howells, D.W. 1026 Experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477.
- Rabinstein, A.A. Update on Treatment of acute ischemic stroke. Contin. Lifelong Learn. Neurol. 2020, 26, 268–286.
- Barthels, D.; Das, H. Current advances in ischemic stroke research and therapies. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866.
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.-P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update. Circulation 2015, 131, 29–39.
- Gibson, C.L. Cerebral ischemic stroke: Is gender important? J. Cereb. Blood Flow Metab. 2013, 33, 1355–1361.
- Siesjo, V.K. Cell damage in the brain: A speculative synthesis. J. Cereb. Blood Flow Metab. 1981, 1, 155–185.
- Katsura, K.; Ekholm, A.; Asplund, B.; Siesjo, B.K. Extracellular pH in the brain during ischemia: Relationship to the severity of lactic acidosis. J. Cereb. Blood Flow Metab. 1991, 11, 597–599.
- Nedergaard, M.; Goldman, S.A.; Desai, S.; Pulsinelli, W.A. Acid-induced death in neurons and glia. J. Neurosci. 1991, 11, 2489–2497.
- Park, J.E.; Jung, S.C.; Kim, H.S.; Suh, J.Y.; Baek, J.H.; Woo, C.W.; Park, B.; Woo, D.C. Amide proton transfer—Weighted MRI can detect tissue acidosis and monitor recovery in a transient middle cerebral artery occlusion model compared with a permanent occlusion model in rats. Eur. Radiol. 2019, 29, 4096–4104.
- Obara, M.; Szeliga, M.; Albrecht, J. Regulation of pH in the mammalian central nervous system under normal and pathological conditions: Facts and hypotheses. Neurochem. Int. 2008, 52, 905–919.
- Siesjö, B.K.; Katsura, K.; Mellergard, P.; Ekholm, A.; Lundgren, J.; Smith, M. Acidosis-related brain damage. Prog. Brain Res. 1993, 96, 1–26.
- Erra Díaz, F.; Dantas, E.; Geffner, J. Unravelling the interplay between extracellular acidosis and immune cells. Mediat. Inflamm. 2018, 2018.
- Rehncrona, S.; Rosen, I.; Siesjo, B. Brain lactic acidosis and ischemic cell damage: I. biochemistry and neurophysiology. J. Cereb. Blood Flow Metab. 1981, 1, 297–311.
- Tóth, O.M.; Menyhárt, Á.; Frank, R.; Hantosi, D.; Farkas, E.; Bari, F. Tissue acidosis associated with ischemic stroke to guide neuroprotective drug delivery. Biology 2020, 9, 460.
- Plum, F. What causes infarction in ischemic brain? The Robert Wartenberg lecture. Neurology 1983, 33, 222–233.
- Swanson, R.A.; Farrell, K.; Simon, R.P. Acidosis causes failure of astrocyte glutamate uptake during hypoxia. J. Cereb. Blood Flow Metab. 1995, 15, 417–424.
- Supuran, C.T. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin. Ther. Pat. 2018, 1744–7674.
- Mishra, C.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565.
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360.
- Nocentini, A.; Angeli, A.; Carta, F.; Winum, J.Y.; Zalubovskis, R.; Carradori, S.; Capasso, C.; Donald, W.A.; Supuran, C.T. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J. Enzyme Inhib. Med. Chem. 2021, 36, 561–580.
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.A.; Capasso, C.; Supuran, C.T. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum—The η-carbonic anhydrases. Bioorgan. Med. Chem. Lett. 2014, 24, 4389–4396.
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181.
- Supuran, C.T. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin. Ther. Pat. 2018, 709–712.
- Aggarwal, M.; Kondeti, B.; McKenna, R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 717–724.
- Berrino, E.; Carta, F. Carbonic anhydrase inhibitors for the treatment of epilepsy and obesity. In Carbonic Anhydrases: Biochemistry and Pharmacology of an Evergreen Pharmaceutical Target; Elsevier: Amsterdam, Netherlands, 2019; pp. 311–329.
- Thiry, A.; Dogne, J.-M.; Supuran, C.T.; Masereel, B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr. Top. Med. Chem. 2007, 7, 855–864.
- Uldall, M.; Botfield, H.; Jansen-Olesen, I.; Sinclair, A.; Jensen, R. Acetazolamide lowers intracranial pressure and modulates the cerebrospinal fluid secretion pathway in healthy rats. Neurosci. Lett. 2017, 645, 33–39.
- Asiedu, M.; Ossipov, M.; Kaila, K.; Price, T. Acetazolamide and midazolam act synergistically to inhibit neuropathic pain. Pain 2010, 148, 302–308.
- Supuran, C.T. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev. Neurother. 2016, 961–968.
- Price, T.; Sheibani, N.; Shah, G. Regulation of high glucose-induced apoptosis of brain pericytes by mitochondrial CA VA: A specific target for prevention of diabetic cerebrovascular pathology. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 929–935.
- Salameh, T.; Shah, G.; Price, T.; Hayden, M.; Banks, W. Blood-brain barrier disruption and neurovascular unit dysfunction in diabetic mice: Protection with the mitochondrial carbonic anhydrase inhibitor topiramate. J. Pharmacol. Exp. Ther. 2016, 359, 452–459.
- Silberstein, S.D. Topiramate in migraine prevention: A 2016 perspective. Headache 2017, 57, 165–178.
- Fossati, S.; Giannoni, P.; Solesio, M.E.; Cocklin, S.L.; Cabrera, E.; Ghiso, J.; Rostagno, A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol. Dis. 2016, 86, 29–40.
- Angiulli, F.; Solesio, M.; Debure, L.; Cejudo, J.; Wisniewski, T.; Fossati, S. Carbonic anydrase inhibitors ameliorate neurovascular dysfunction in a mouse model of cerebral amyloid angiopathy. Alzheimer’s Dement. 2018, 14.
- Provensi, G.; Carta, F.; Nocentini, A.; Supuran, C.T.; Casamenti, F.; Passani, M.B.; Fossati, S. A new kid on the block? Carbonic anhydrases as possible new targets in alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 4724.
- Velísek, L.; Moshé, S.; Stanton, P. Resistance of hippocampal synaptic transmission to hypoxia in carbonic anhydrase II-deficient mice. Brain Res. 1995, 671, 245–253.
- Kniep, E.M.; Roehlecke, C.; Özkucur, N.; Steinberg, A.; Reber, F.; Knels, L.; Funk, R.H.W. Inhibition of apoptosis and reduction of intracellular pH decrease in retinal neural cell cultures by a blocker of carbonic anhydrase. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1185–1192.
- Wykoff, C.C.; Beasley, N.J.P.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083.
- Stiehl, D.P.; Wirthner, R.; Köditz, J.; Spielmann, P.; Camenisch, G.; Wenger, R.H. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels: Evidence for an autoregulatory oxygen-sensing system. J. Biol. Chem. 2006, 281, 23482–23491.
- Pettersen, E.O.; Ebbesen, P.; Gieling, R.G.; Williams, K.J.; Dubois, L.; Lambin, P.; Ward, C.; Meehan, J.; Kunkler, I.H.; Langdon, S.P.; et al. Targeting tumour hypoxia to prevent cancer metastasis. From biology, biosensing and technology to drug development: The METOXIA consortium. J. Enzyme Inhib. Med. Chem. 2015, 30, 689–721.
- Svichar, N.; Esquenazi, S.; Waheed, A.; Sly, W.S.; Chesler, M. Functional demonstration of surface carbonic anhydrase IV activity on rat astrocytes. Glia 2006, 53, 241–247.
- Tong, C.; Brion, L.; Suarez, C.; Chesler, M. Interstitial carbonic anhydrase (CA) activity in brain is attributable to membrane-bound CA type IV. J. Neurosci. 2000, 20, 8247–8253.
- Deitmer, J.; Theparambil, S.; Ruminot, I.; Noor, S.; Becker, H. Energy dynamics in the brain: Contributions of astrocytes to Metabolism and pH homeostasis. Front. Neurosci. 2019, 13, 1301.
- Bélanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295.
- Giffard, R.; Monyer, H.; Choi, D. Selective vulnerability of cultured cortical glia to injury by extracellular acidosis. Brain Res. 1990, 530, 138–141.
- Cannizzaro, C.; Monastero, R.; Vacca, M.; Martire, M. [3H]-DA release evoked by low pH medium and internal H+ accumulation in rat hypothalamic synaptosomes: Involvement of calcium ions. Neurochem. Int. 2003, 43, 9–17.
- Pittaluga, A.; Segantini, D.; Feligioni, M.; Raiteri, M. Extracellular protons differentially potentiate the responses of native AMPA receptor subtypes regulating neurotransmitter release. Br. J. Pharmacol. 2005, 144, 293–299.
- Beppu, K.; Sasaki, T.; Tanaka, K.; Yamanaka, A.; Fukazawa, Y.; Shigemoto, R.; Matsui, K. Optogenetic countering of glial acidosis suppresses glial glutamate release and ischemic brain damage. Neuron 2014, 81, 314–320.
- Somjen, G.G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 2001, 81, 1065–1096.
- Rasmussen, J.K.; Boedtkjer, E. Carbonic anhydrase inhibitors modify intracellular pH transients and contractions of rat middle cerebral arteries during CO 2 /HCO 3– fluctuations. J. Cereb. Blood Flow Metab. 2018, 38, 492–505.
- Lisk, C.; McCord, J.; Bose, S.; Sullivan, T.; Loomis, Z.; Nozik-Grayck, E.; Schroeder, T.; Hamilton, K.; Irwin, D.C. Nrf2 activation: A potential strategy for the prevention of acute mountain sickness. Free Radic. Biol. Med. 2013, 63, 264–273.
- Gao, B.B.; Clermont, A.; Rook, S.; Fonda, S.J.; Srinivasan, V.J.; Wojtkowski, M.; Fujimoto, J.G.; Avery, R.L.; Arrigg, P.G.; Bursell, S.E.; et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat. Med. 2007, 13, 181–188.
- Guo, F.; Hua, Y.; Wang, J.; Keep, R.; Xi, G. Inhibition of carbonic anhydrase reduces brain injury after intracerebral hemorrhage. Transl. Stroke Res. 2012, 3, 130–137.
- Hladky, S.B.; Barrand, M.A. Fluid and ion transfer across the blood-brain and blood-cerebrospinal fluid barriers; a comparative account of mechanisms and roles. Fluids Barriers CNS 2016, 13.
- Kamegawa, A.; Hiroaki, Y.; Tani, K.; Fujiyoshi, Y. Two-dimensional crystal structure of aquaporin-4 bound to the inhibitor acetazolamide. Reprod. Syst. Sex. Disord. 2016, 65, 177–184.
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 and brain edema. Pediatr. Nephrol. 2007, 22, 778–784.
- Supuran, C.T.; Altamini, A.; Carta, F. Carbonic anhydrase inhibition and the management of glaucoma: A literature and patent review 2013–2019. Expert Opin. Ther. Pat. 2019, 29, 781–792.
- Bua, S.; Nocentini, A.; Supuran, C.T. Carbonic anhydrase inhibitors as diuretics. In Carbonic Anhydrases: Biochemistry and Pharmacology of an Evergreen Pharmaceutical Target; Elsevier: Amsterdam, Netherlands, 2019; pp. 287–309.
- Davis, C.; Hackett, P. Advances in the prevention and treatment of high altitude illness. Emerg. Med. Clin. N. Am. 2017, 35, 241–260.
- Dostovic, Z.; Dostovic, E.; Smajlovic, D.; Ibrahimagic, O.; Avdic, L. Brain edema after ischaemic stroke. Med. Arch. 2016, 70, 339–341.
- Klatzo, I. Brain oedema following brain ischaemia and the influence of therapy. Br. J. Anaesth. 1985, 57, 18–22.
- Domoki, F.; Zimmermann, A.; Tóth-Szuki, V.; Busija, D.; Bari, F. Acetazolamide induces indomethacin and ischaemia-sensitive pial arteriolar vasodilation in the piglet. Acta Paediatr. Int. J. Paediatr. 2008, 97, 280–284.
- Vorstrup, S.; Henriksen, L.; Paulson, O.B. Effect of acetazolamide on cerebral blood flow and cerebral metabolic rate for oxygen. J. Clin. Investig. 1984, 74, 1634–1639.
- Tuettenberg, J.; Heimann, A.; Kempski, O. Nitric oxide modulates cerebral blood flow stimulation by acetazolamide in the rat cortex: A laser doppler scanning study. Neurosci. Lett. 2001, 315, 65–68.
- Kiss, B.; Dallinger, S.; Findl, O.; Rainer, G.; Eichler, H.G.; Schmetterer, L. Acetazolamide-induced cerebral and ocular vasodilation in humans is independent of nitric oxide. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1999, 276.
- Pickkers, P.; Hughes, A.D.; Russel, F.G.M.; Thien, T.; Smits, P. In vivo evidence for KCa channel opening properties of acetazolamide in the human vasculature. Br. J. Pharmacol. 2001, 132, 443–450.
- Yu, H.; Qi, G.L.; Wang, J.; Chen, L.; Deng, Z.; Zhao, Y.S.; Lei, S.S.; Zhu, X.Q. Aquaporin 4 inhibition decreased synthesis of cytokines by acetazolamide in the hippocampus of rats with pentrazol-induced chronic epilepsy. Genet. Mol. Res. 2016, 15.
- Wang, C.; Wang, R.; Xie, H.; Sun, Y.; Tao, R.; Liu, W.; Li, W.; Lu, H.; Jia, Z. Effect of acetazolamide on cytokines in rats exposed to high altitude. Cytokine 2016, 83, 110–117.

