 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zarema Albakova | + 5080 word(s) | 5080 | 2021-05-19 07:40:38 | | | |
| 2 | Vivi Li | Meta information modification | 5080 | 2021-05-19 10:51:40 | | |
Video Upload Options
The 70-kDa heat shock proteins (HSP70s) are abundantly present in cancer, providing malignant cells selective advantage by suppressing multiple apoptotic pathways, regulating necrosis, bypassing cellular senescence program, interfering with tumor immunity, promoting angiogenesis and supporting metastasis. This direct involvement of HSP70 in most of the cancer hallmarks explains the phenomenon of cancer “addiction” to HSP70, tightly linking tumor survival and growth to the HSP70 expression. HSP70 operates in different states through its catalytic cycle, suggesting that it can multi-function in malignant cells in any of these states. Clinically, tumor cells intensively release HSP70 in extracellular microenvironment, resulting in diverse outcomes for patient survival. Given its clinical significance, small molecule inhibitors were developed to target different sites of the HSP70 machinery. Furthermore, several HSP70-based immunotherapy approaches were assessed in clinical trials.
1. Introduction
2. The HSP70 Machinery
2.1. HSP70 Structure
2.2. HSP70 Functional Cycle
2.3. The Internal HSP70 Network
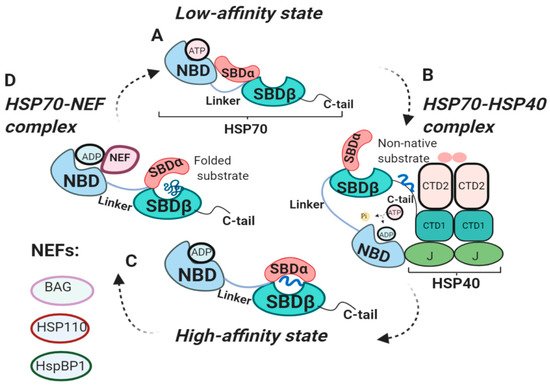
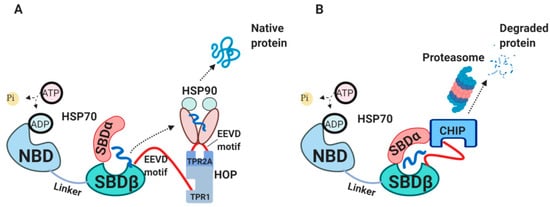
2.4. The External HSP70 Network
3. Diverse Functions of HSP70 in the Hallmarks of Cancer
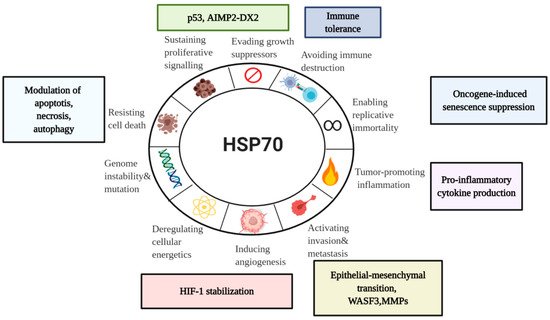
3.1. HSP70 and Tumor Immunity
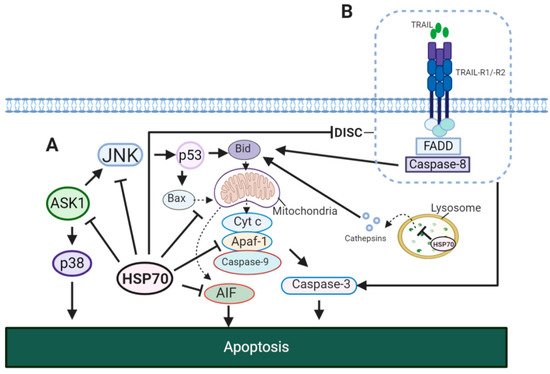
3.2. HSP70 and Tumor Resistance to Cell Death
3.3. HSP70 and Senescence Program
3.4. HSP70 in Sustained Proliferation
3.5. HSP70 in Angiogenesis
3.6. HSP70 in Metastasis
4. HSP70 Therapies Targeting Cancer
4.1. Small Molecule Inhibitors of HSP70 Cycle
| HSP70-Targeting Molecules | Effects | Refs. | |
|---|---|---|---|
| SBD-targeting inhibitors | PES (Pifithrin-µ) | Suppressed tumor development in a mouse model of Myc-lymphoma. | [170] |
| ADD70 | Increased sensitivity of colon cancer and melanoma cells to apoptosis; showed antimetastatic effects in vivo. | [171] | |
| Acridizinium derivative 1 | Induced apoptosis in HeLa cells. | [172] | |
| NBD-targeting inhibitors | Synthetic peptide P17 | Decreased melanoma growth in vivo. | [173] |
| JG-98 inhibitor (MKT-077 analog) |
Decreased tumor growth in MCF7 xenograft model. | [164] | |
| VER-155008 | Inhibited proliferation of human breast and colon cancer cell lines. | [168] | |
| YK-5 | Induced apoptosis and degradation of HSP70/HSP90 client proteins (HER2,Raf-1, Akt kinases) in breast cancer cells. | [174] | |
| Apoptozole | Induced apoptosis in ovarian, colon and lung cancer cell lines. | [175][176] | |
| HSP70-HSP40 complex inhibitors | MAL3-101 | Inhibited proliferation of multiple myeloma cells derived from patients. | [177] |
| DMT3132 (MAL3-101 analog) |
Reduced proliferation in breast cancer cells. | [178][179] | |
| Myricetin | Inhibited tumor growth in pancreatic cancer. | [180] | |
| HSP70-NEF complex inhibitors | YM-1 | Inhibited tumor growth in mammary and melanoma xenograft models. | [119] |
| Allosteric HSP70 inhibitors | HS-72 | Inhibited growth in HER2-positive breast cancel model. | [181] |
4.2. Other HSP70 Inhibitors
4.3. HSP70-Based Therapies in Cancer Clinical Trials
| HSP70-Based Therapies | Condition | Clinical Trial Phase/Case Study | Refs. |
|---|---|---|---|
| Autologous HSP70-peptide complex in combination with imatinib mesylate | Chronic myeloid leukemia | Phase I | [198] |
| Autologous HSP70-peptide complex (AG-858) in combination with Gleevec | Chronic Myelogenous Leukemia | Phase II | [199] |
| Autologous peripheral blood mononuclear cells (PBMC) pre-activated with TKD peptide/IL-2 | Colon carcinoma | Case study | [193] |
| Autologous NK cells pre-activated with TKD and IL-2 following radiochemotherapy | Advanced colorectal carcinoma and non- small cell lung carcinoma (NSCLC) | Phase I; Phase II | [71][194] |
| DNA vaccine expressing HPV16 E7 mutant form and HSP70 | Cervical intraepithelial neoplasia | Phase I | [195] |
| Dendritic cells transfected with HSP70 mRNA | Hepatocellular carcinoma | Phase I | [196] |
| Autologous NK cells pre-treated with TKD/IL-2 in combination with radiochemotherapy and nivolumab (PD-1 antibody) | NSCLC stage IIIb | Case study | [197] |
| Intratumoral injection of recombinant oncolytic type 2 adenovirus overexpressing HSP70 (H103) | Advanced solid tumors | Phase I | [200] |
References
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Cell. Mol. Life Sci. 1962, 18, 571–573.
- Tissières, A.; Mitchell, H.K.; Tracy, U.M. Protein synthesis in salivary glands of Drosophila melanogaster: Relation to chromosome puffs. J. Mol. Biol. 1974, 84, 389–398.
- Moran, L.; Mirault, M.-E.; Arrigo, A.P.; Goldschmidt-Clermont, M.; Tissières, A. Heat shock of Drosophila melanogaster induces the synthesis of new messenger RNAs and proteins. Philos. Trans. R. Soc. B Biol. Sci. 1978, 283, 391–406.
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710.
- Beere, H. Stress management—Heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001, 11, 6–10.
- Yang, X.; Wang, J.; Zhou, Y.; Wang, Y.; Wang, S.; Zhang, W.-Y. Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells. Cancer Lett. 2012, 321, 137–143.
- Pelham, H. Hsp70 accelerates the recovery of nucleolar morphology after heat shock. EMBO J. 1984, 3, 3095–3100.
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680.
- Hunt, C.; Morimoto, R.I. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proc. Natl. Acad. Sci. USA 1985, 82, 6455–6459.
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677.
- Gupta, R.S.; Singh, B. Phylogenetic analysis of 70 kD heat shock protein sequences suggests a chimeric origin for the eukaryotic cell nucleus. Curr. Biol. 1994, 4, 1104–1114.
- Mosser, D.D.; Morimoto, R.I. Molecular chaperones and the stress of oncogenesis. Oncogene 2004, 23, 2907–2918.
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103.
- Calderwood, S.; Khaleque, A.; Sawyer, U.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172.
- Kampinga, H.H.; Craig, E. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592.
- Arakawa, A.; Handa, N.; Ohsawa, N.; Shida, M.; Kigawa, T.; Hayashi, F.; Shirouzu, M.; Yokoyama, S. The C-Terminal BAG Domain of BAG5 Induces Conformational Changes of the Hsp70 Nucleotide- Binding Domain for ADP-ATP Exchange. Structure 2010, 18, 309–319.
- Arakawa, A.; Handa, N.; Shirouzu, M.; Yokoyama, S. Biochemical and structural studies on the high affinity of Hsp70 for ADP. Protein Sci. 2011, 20, 1367–1379.
- Cheeseman, M.; Westwood, I.M.; Barbeau, O.; Rowlands, M.; Dobson, S.; Jones, A.M.; Jeganathan, F.; Burke, R.; Kadi, N.; Workman, P.; et al. Exploiting Protein Conformational Change to Optimize Adenosine-Derived Inhibitors of HSP70. J. Med. Chem. 2016, 59, 4625–4636.
- Gao, X.-C.; Zhou, C.-J.; Zhou, Z.-R.; Wu, M.; Cao, C.-Y.; Hu, H.-Y. The C-terminal Helices of Heat Shock Protein 70 Are Essential for J-domain Binding and ATPase Activation*. J. Biol. Chem. 2012, 287, 6044–6052.
- Hassan, A.Q.; Kirby, C.A.; Zhou, W.; Schuhmann, T.; Kityk, R.; Kipp, D.R.; Baird, J.; Chen, J.; Chen, Y.; Chung, F.; et al. The Novolactone Natural Product Disrupts the Allosteric Regulation of Hsp70. Chem. Biol. 2015, 22, 87–97.
- Jones, A.M.; Westwood, I.M.; Osborne, J.D.; Matthews, T.P.; Cheeseman, M.; Rowlands, M.G.; Jeganathan, F.; Burke, R.; Lee, D.; Kadi, N.; et al. A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms. Sci. Rep. 2016, 6, 34701.
- Lim, S.; Cho, H.Y.; Kim, D.G.; Roh, Y.; Son, S.-Y.; Mushtaq, A.U.; Kim, M.; Bhattarai, D.; Sivaraman, A.; Lee, Y.; et al. Targeting the interaction of AIMP2-DX2 with HSP70 suppresses cancer development. Nat. Methods 2019, 16, 31–41.
- Osipiuk, J.; Walsh, M.A.; Freeman, B.C.; Morimoto, R.I.; Joachimiak, A. Structure of a new crystal form of human Hsp70 ATPase domain. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999, 55, 1105–1107.
- Ravalin, M.; Theofilas, P.; Basu, K.; Opoku-Nsiah, K.A.; Assimon, V.A.; Medina-Cleghorn, D.; Chen, Y.-F.; Bohn, M.F.; Arkin, M.; Grinberg, L.T.; et al. Specificity for latent C termini links the E3 ubiquitin ligase CHIP to caspases. Nat. Methods 2019, 15, 786–794.
- Shida, M.; Arakawa, A.; Ishii, R.; Kishishita, S.; Takagi, T.; Kukimoto-Niino, M.; Sugano, S.; Tanaka, A.; Shirouzu, M.; Yokoyama, S. Direct inter-subdomain interactions switch between the closed and open forms of the Hsp70 nucleotide-binding domain in the nucleotide-free state. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 223–232.
- Sriram, M.; Osipiuk, J.; Freeman, B.; Morimoto, R.; Joachimiak, A. Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure 1997, 5, 403–414.
- Umehara, K.; Hoshikawa, M.; Tochio, N.; Tate, S.-I. Substrate Binding Switches the Conformation at the Lynchpin Site in the Substrate-Binding Domain of Human Hsp70 to Enable Allosteric Interdomain Communication. Molecules 2018, 23, 528.
- Zhang, P.; Leu, J.I.-J.; Murphy, M.E.; George, D.L.; Marmorstein, R. Crystal Structure of the Stress-Inducible Human Heat Shock Protein 70 Substrate-Binding Domain in Complex with Peptide Substrate. PLoS ONE 2014, 9, e103518.
- Murphy, M.E. The HSP70 family and cancer. Carcinogen 2013, 34, 1181–1188.
- Ha, J.-H.; McKay, D.B. Kinetics of Nucleotide-Induced Changes in the Tryptophan Fluorescence of the Molecular Chaperone Hsc70 and Its Subfragments Suggest the ATP-Induced Conformational Change Follows Initial ATP Binding. Biochemistry 1995, 34, 11635–11644.
- Palleros, D.R.; Raid, K.L.; Shi, L.; Welch, W.J.; Fink, A.L. ATP-induced protein Hsp70 complex dissociation requires K+ but not ATP hydrolysis. Nature 1993, 365, 664–666.
- Schmid, D.; Baici, A.; Gehring, H.; Christen, P. Kinetics of molecular chaperone action. Science 1994, 263, 971–973.
- Prasad, K.; Heuser, J.; Eisenberg, E.; Greene, L. Complex formation between clathrin and uncoating ATPase. J. Biol. Chem. 1994, 269, 6931–6939.
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–580.
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457.
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684.
- Suzuki, H.; Noguchi, S.; Arakawa, H.; Tokida, T.; Hashimoto, M.; Satow, Y. Peptide-Binding Sites As Revealed by the Crystal Structures of the Human Hsp40 Hdj1 C-Terminal Domain in Complex with the Octapeptide from Human Hsp70. Biochemistry 2010, 49, 8577–8584.
- Jiang, Y.; Rossi, P.; Kalodimos, C.G. Structural basis for client recognition and activity of Hsp40 chaperones. Science 2019, 365, 1313–1319.
- Craig, E.; Huang, P.; Aron, R.; Andrew, A. The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Ergeb. der Physiol. 2006, 156.
- Hageman, J.; Kampinga, H.H. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones 2008, 14, 1–21.
- Diamant, S.; Goloubinoff, P. Temperature-Controlled Activity of DnaK−DnaJ−GrpE Chaperones: Protein-Folding Arrest and Recovery during and after Heat Shock Depends on the Substrate Protein and the GrpE Concentration†. Biochemistry 1998, 37, 9688–9694.
- Mitra, A.; Shevde, L.A.; Samant, R.S. Multi-faceted role of HSP40 in cancer. Clin. Exp. Metastasis 2009, 26, 559–567.
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10.
- Brodsky, J.L.; Bracher, A. Nucleotide Exchange Factors for HSP70 Molecular Chaperones. In Networking of Chaperones by Co-Chaperones; Molecular Biology Intelligence Unit; Springer: New York, NY, USA, 2007; pp. 1–12.
- Li, Z.; Hartl, F.U.; Bracher, A. Structure and function of Hip, an attenuator of the Hsp70 chaperone cycle. Nat. Struct. Mol. Biol. 2013, 20, 929–935.
- Wang, A.M.; Miyata, Y.; Klinedinst, S.; Peng, H.-M.; Chua, J.P.; Komiyama, T.; Li, X.; Morishima, Y.; Merry, D.E.; Pratt, W.B.; et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Methods 2012, 9, 112–118.
- Rousaki, A.; Miyata, Y.; Jinwal, U.K.; Dickey, C.A.; Gestwicki, J.E.; Zuiderweg, E.R.P. Allosteric Drugs: The Interaction of Antitumor Compound MKT-077 with Human Hsp70 Chaperones. J. Mol. Biol. 2011, 411, 614–632.
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.; Moarefi, I. Structure of TPR Domain–Peptide Complexes. Cell 2000, 101, 199–210.
- Smith, D.F.; Sullivan, W.P.; Marion, T.N.; Zaitsu, K.; Madden, B.; McCormick, D.J.; O Toft, D. Identification of a 60-kilodalton stress-related protein, p60, which interacts with hsp90 and hsp70. Mol. Cell. Biol. 1993, 13, 869–876.
- Honoré, B.; Leffers, H.; Madsen, P.; Rasmussen, H.H.; Vandekerckhove, J.; E Celis, J. Molecular cloning and expression of a transformation-sensitive human protein containing the TPR motif and sharing identity to the stress-inducible yeast protein STI1. J. Biol. Chem. 1992, 267, 8485–8491.
- Stankiewicz, M.; Nikolay, R.; Rybin, V.; Mayer, M.P. CHIP participates in protein triage decisions by preferentially ubiquitinating Hsp70-bound substrates. FEBS J. 2010, 277, 3353–3367.
- Höhfeld, J.; Cyr, D.M.; Patterson, C. From the cradle to the grave: Molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001, 2, 885–890.
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Höhfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nature 2000, 3, 93–96.
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nature 2000, 3, 100–105.
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.-Y.; Patterson, C. Identification of CHIP, a Novel Tetratricopeptide Repeat-Containing Protein That Interacts with Heat Shock Proteins and Negatively Regulates Chaperone Functions. Mol. Cell. Biol. 1999, 19, 4535–4545.
- Zhang, H.; Amick, J.; Chakravarti, R.; Santarriaga, S.; Schlanger, S.; McGlone, C.; Dare, M.; Nix, J.C.; Scaglione, K.M.; Stuehr, D.J.; et al. A Bipartite Interaction between Hsp70 and CHIP Regulates Ubiquitination of Chaperoned Client Proteins. Structure 2015, 23, 472–482.
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76.
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell. Biol. 2017, 18, 345–360.
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365.
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.S.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Srivastava, P.K. Interaction of Heat Shock Proteins with Peptides and Antigen Presenting Cells: Chaperoning of the Innate and Adaptive Immune Responses. Annu. Rev. Immunol. 2002, 20, 395–425.
- Blachere, N.E.; Li, Z.; Chandawarkar, R.Y.; Suto, R.; Jaikaria, N.S.; Basu, S.; Udono, H.; Srivastava, P.K. Heat Shock Protein–Peptide Complexes, Reconstituted In Vitro, Elicit Peptide-specific Cytotoxic T Lymphocyte Response and Tumor Immunity. J. Exp. Med. 1997, 186, 1315–1322.
- Multhoff, G.; Botzler, C.; Wiesnet, M.; Müller, E.; Meier, T.; Wilmanns, W.; Issels, R.D. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 1995, 61, 272–279.
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247.
- Stangl, S.; Gross, C.; Pockley, A.G.; Asea, A.; Multhoff, G. Influence of Hsp70 and HLA-E on the killing of leukemic blasts by cytokine/Hsp70 peptide-activated human natural killer (NK) cells. Cell Stress Chaperones 2008, 13, 221–230.
- Multhoff, G.; Pfister, K.; Gehrmann, M.; Hantschel, M.; Gross, C.; Hafner, M.; Hiddemann, W. A 14-mer Hsp70 peptide stimulates natural killer (NK) cell activity. Cell Stress Chaperons 2001, 6, 337–344.
- Multhoff, G.; Botzler, C.; Jennen, L.; Schmidt, J.; Ellwart, J.; Issels, R. Heat shock protein 72 on tumor cells: A recognition structure for natural killer cells. J. Immunol. 1997, 158, 4341–4350.
- Multhoff, G. Heat shock protein 70 (Hsp70) stimulates proliferation and cytolytic activity of natural killer cells. Exp. Hematol. 1999, 27, 1627–1636.
- Specht, H.M.; Ahrens, N.; Blankenstein, C.; Duell, T.; Fietkau, R.; Gaipl, U.S.; Günther, C.; Gunther, S.; Habl, G.; Hautmann, H.; et al. Heat Shock Protein 70 (Hsp70) Peptide Activated Natural Killer (NK) Cells for the Treatment of Patients with Non-Small Cell Lung Cancer (NSCLC) after Radiochemotherapy (RCTx) – From Preclinical Studies to a Clinical Phase II Trial. Front. Immunol. 2015, 6.
- Hromadníková, I.; Li, S.; Kotlabova, K.; Dickinson, A.M. Influence of In Vitro IL-2 or IL-15 Alone or in Combination with Hsp 70 Derived 14-Mer Peptide (TKD) on the Expression of NK Cell Activatory and Inhibitory Receptors on Peripheral Blood T Cells, B Cells and NKT Cells. PLoS ONE 2016, 11, e0151535.
- Rückert, M.; Deloch, L.; Fietkau, R.; Frey, B.; Hecht, M.; Gaipl, U.S. Immune modulatory effects of radiotherapy as basis for well-reasoned radioimmunotherapies. Strahlenther. und Onkol. 2018, 194, 509–519.
- Günther, S.; Ostheimer, C.; Stangl, S.; Specht, H.M.; Mozes, P.; Jesinghaus, M.; Vordermark, D.; Combs, S.E.; Peltz, F.; Jung, M.P.; et al. Correlation of Hsp70 Serum Levels with Gross Tumor Volume and Composition of Lymphocyte Subpopulations in Patients with Squamous Cell and Adeno Non-Small Cell Lung Cancer. Front. Immunol. 2015, 6, 128.
- Ostheimer, C.; Gunther, S.; Bache, M.; Vordermark, D.; Multhoff, G. Dynamics of Heat Shock Protein 70 Serum Levels As a Predictor of Clinical Response in Non-Small-Cell Lung Cancer and Correlation with the Hypoxia-Related Marker Osteopontin. Front. Immunol. 2017, 8.
- A Daniels, G.; Sanchez-Perez, L.; Diaz, R.M.; Kottke, T.; Thompson, J.; Lai, M.; Gough, M.; Karim, M.; Bushell, A.; Chong, H.; et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat. Biotechnol. 2004, 22, 1125–1132.
- Breloer, M.; Fleischer, B.; Von Bonin, A. In vivo and in vitro activation of T cells after administration of Ag-negative heat shock proteins. J. Immunol. 1999, 162.
- Rothammer, A.; Sage, E.K.; Werner, C.; Combs, S.E.; Multhoff, G. Increased heat shock protein 70 (Hsp70) serum levels and low NK cell counts after radiotherapy - potential markers for predicting breast cancer recurrence? Radiat. Oncol. 2019, 14, 78.
- Pfister, K.; Radons, J.; Busch, R.; Tidball, J.G.; Pfeifer, M.; Freitag, L.; Feldmann, H.-J.; Milani, V.; Issels, R.; Multhoff, G. Patient survival by Hsp70 membrane phenotype. Cancer 2007, 110, 926–935.
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846.
- Wachstein, J.; Tischer, S.; Figueiredo, C.; Limbourg, A.; Falk, C.; Immenschuh, S.; Blasczyk, R.; Eiz-Vesper, B. HSP70 Enhances Immunosuppressive Function of CD4+CD25+FoxP3+ T Regulatory Cells and Cytotoxicity in CD4+CD25− T Cells. PLoS ONE 2012, 7, e51747.
- Asea, A.; Kraeft, S.-K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442.
- Asea, A. Novel Signal Transduction Pathway Utilized by Extracellular HSP70. Role of Toll-Like Receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034.
- Asea, A. Initiation of the Immune Response by Extracellular Hsp72: Chaperokine Activity of Hsp72. Curr. Immunol. Rev. 2006, 2, 209–215.
- Asea, A. Chaperokine-induced signal transduction pathways. Exerc. Immunol. Rev. 2003, 9, 25–33.
- Martine, P.; Chevriaux, A.; Derangère, V.; Apetoh, L.; Garrido, C.; Ghiringhelli, F.; Rebe, C. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 2019, 10, 256.
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158.
- Yashin, D.V.; Romanova, E.A.; Ivanova, O.K.; Sashchenko, L.P. The Tag7–Hsp70 cytotoxic complex induces tumor cell necroptosis via permeabilisation of lysosomes and mitochondria. Biochimie 2016, 123, 32–36.
- Balogi, Z.; Multhoff, G.; Jensen, T.K.; Lloyd-Evans, E.; Yamashima, T.; Jäättelä, M.; Harwood, J.L.; Vígh, L.; Multhoff, G. Hsp70 interactions with membrane lipids regulate cellular functions in health and disease. Prog. Lipid Res. 2019, 74, 18–30.
- Sashchenko, L.P.; Dukhanina, E.A.; Yashin, D.V.; Shatalov, Y.V.; Romanova, E.A.; Korobko, E.V.; Demin, A.V.; Lukyanova, T.I.; Kabanova, O.D.; Khaidukov, S.V.; et al. Peptidoglycan Recognition Protein Tag7 Forms a Cytotoxic Complex with Heat Shock Protein 70 in Solution and in Lymphocytes. J. Biol. Chem. 2003, 279, 2117–2124.
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.-P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471.
- Bausero, M.A.; Gastpar, R.; Multhoff, G.; Asea, A. Alternative Mechanism by which IFN-γ Enhances Tumor Recognition: Active Release of Heat Shock Protein 721. J. Immunol. 2005, 175, 2900–2912.
- De Vita, F.; Orditura, M.; Galizia, G.; Romano, C.; Infusino, S.; Auriemma, A.; Lieto, E.; Catalano, G. Serum interleukin-10 levels in patients with advanced gastrointestinal malignancies. Cancer 1999, 86, 1936–1943.
- Lebrecht, A.; Hefler, L.; Tempfer, C.B.; Koelbl, H. Serum Cytokine Concentrations in Patients with Cervical Cancer: Interleukin-4, Interferon-γ, and Monocyte Chemoattractant Protein-1. Gynecol. Oncol. 2001, 83, 170–171.
- Udono, H.; Srivastava, P.K. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J. Immunol. 1994, 152, 5398–5403.
- Weng, D.; Calderwood, S.K.; Gong, J. Preparation of a heat-shock protein 70-based vaccine from DC-tumor fusion cells. Breast Cancer 2011, 787, 255–265.
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35.
- Calderwood, S.K.; Murshid, A. Molecular Chaperone Accumulation in Cancer and Decrease in Alzheimer’s Disease: The Potential Roles of HSF1. Front. Neurosci. 2017, 11, 192.
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Denis-Larose, C.; Massie, B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol. 1997, 17, 5317–5327.
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jäättelä, M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876.
- Gao, Y.; Han, C.; Huang, H.; Xin, Y.; Xu, Y.; Luo, L.; Yin, Z. Heat shock protein 70 together with its co-chaperone CHIP inhibits TNF-α induced apoptosis by promoting proteasomal degradation of apoptosis signal-regulating kinase1. Apoptosis 2010, 15, 822–833.
- Tournier, C. Requirement of JNK for Stress- Induced Activation of the Cytochrome c-Mediated Death Pathway. Science 2000, 288, 870–874.
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251.
- Sax, J.K.; Fei, P.; Murphy, M.E.; Bernhard, E.; Korsmeyer, S.J.; El-Deiry, W.S. BID regulation by p53 contributes to chemosensitivity. Nature 2002, 4, 842–849.
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jäättelä, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nature 2001, 3, 839–843.
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in Bcr-Abl–mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255.
- Gabai, V.; Mabuchi, K.; Mosser, D.D.; Sherman, M. Hsp72 and Stress Kinase c-jun N-Terminal Kinase Regulate the Bid-Dependent Pathway in Tumor Necrosis Factor-Induced Apoptosis. Mol. Cell. Biol. 2002, 22, 3415–3424.
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Høyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jäättelä, M. Heat Shock Protein 70 Promotes Cell Survival by Inhibiting Lysosomal Membrane Permeabilization. J. Exp. Med. 2004, 200, 425–435.
- Bivik, C.; Rosdahl, I.; Öllinger, K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogen 2006, 28, 537–544.
- Daugaard, M.; Kirkegaard-Sorensen, T.; Ostenfeld, M.S.; Aaboe, M.; Hoyer-Hansen, M.; Orntoft, T.F.; Rohde, M.; Jäättelä, M. Lens Epithelium-Derived Growth Factor Is an Hsp70-2 Regulated Guardian of Lysosomal Stability in Human Cancer. Cancer Res. 2007, 67, 2559–2567.
- Park, S.-H.; Baek, K.-H.; Shin, I.; Shin, I. Subcellular Hsp70 Inhibitors Promote Cancer Cell Death via Different Mechanisms. Cell Chem. Biol. 2018, 25, 1242–1254.e8.
- Yaglom, J.A.; Ekhterae, D.; Gabai, V.; Sherman, M. Regulation of Necrosis of H9c2 Myogenic Cells upon Transient Energy Deprivation. J. Biol. Chem. 2003, 278, 50483–50496.
- Karsch-Bluman, A.; Feiglin, A.; Arbib, E.; Stern, T.; Shoval, H.; Schwob, O.; Berger, M.; Benny, O. Tissue necrosis and its role in cancer progression. Oncogene 2018, 38, 1920–1935.
- Stankiewicz, A.R.; Lachapelle, G.; Foo, C.P.Z.; Radicioni, S.M.; Mosser, D.D. Hsp70 Inhibits Heat-induced Apoptosis Upstream of Mitochondria by Preventing Bax Translocation. J. Biol. Chem. 2005, 280, 38729–38739.
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nature 2000, 2, 469–475.
- Yaglom, J.A.; Gabai, V.; Sherman, M. High Levels of Heat Shock Protein Hsp72 in Cancer Cells Suppress Default Senescence Pathways. Cancer Res. 2007, 67, 2373–2381.
- Gabai, V.; Yaglom, J.A.; Waldman, T.; Sherman, M.Y. Heat Shock Protein Hsp72 Controls Oncogene-Induced Senescence Pathways in Cancer Cells. Mol. Cell. Biol. 2008, 29, 559–569.
- Meng, L.; Hunt, C.; Yaglom, J.A.; Gabai, V.; Sherman, M.Y. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene 2011, 30, 2836–2845.
- Colvin, T.A.; Gabai, V.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740.
- Meng, L.; Gabai, V.; Sherman, M.Y. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 2010, 29, 5204–5213.
- Oda, T.; Sekimoto, T.; Kurashima, K.; Fujimoto, M.; Nakai, A.; Yamashita, T. Acute HSF1 depletion induces cellular senescence through the MDM2-p53-p21 pathway in human diploid fibroblasts. J. Cell Sci. 2018, 131, jcs210724.
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-Mediated Regulation of the Conformation of p53 DNA Binding Domain and p53 Cancer Variants. Mol. Cell 2019, 74, 831–843.e4.
- Dahiya, V.; Agam, G.; Lawatscheck, J.; Rutz, D.A.; Lamb, D.C.; Buchner, J. Coordinated Conformational Processing of the Tumor Suppressor Protein p53 by the Hsp70 and Hsp90 Chaperone Machineries. Mol. Cell 2019, 74, 816–830.e7.
- Han, J.M.; Park, B.-J.; Park, S.G.; Oh, Y.S.; Choi, S.J.; Lee, S.W.; Hwang, S.-K.; Chang, S.-H.; Cho, M.-H.; Kim, S. AIMP2/p38, the scaffold for the multi-tRNA synthetase complex, responds to genotoxic stresses via p53. Proc. Natl. Acad. Sci. USA 2008, 105, 11206–11211.
- Huang, L.E.; Bunn, H.F. Hypoxia-inducible Factor and Its Biomedical Relevance. J. Biol. Chem. 2003, 278, 19575–19578.
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015.
- Gordan, J.D.; Simon, M.C. Hypoxia-inducible factors: Central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 2007, 17, 71–77.
- Zhou, J.; Schmid, T.; Frank, R.; Brüne, B. PI3K/Akt Is Required for Heat Shock Proteins to Protect Hypoxia-inducible Factor 1 from pVHL-independent Degradation. J. Biol. Chem. 2004, 279, 13506–13513.
- Gabai, V.; Meng, L.; Kim, G.; Mills, T.A.; Benjamin, I.J.; Sherman, M.Y. Heat Shock Transcription Factor Hsf1 Is Involved in Tumor Progression via Regulation of Hypoxia-Inducible Factor 1 and RNA-Binding Protein HuR. Mol. Cell. Biol. 2012, 32, 929–940.
- De Silanes, I.L.; Fan, J.; Yang, X.; Zonderman, A.B.; Potapova, O.; Pizer, E.S.; Gorospe, M. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 2003, 22, 7146–7154.
- Kim, T.-K.; Na, H.-J.; Lee, W.R.; Jeoung, M.H.; Lee, S. Heat shock protein 70-1A is a novel angiogenic regulator. Biochem. Biophys. Res. Commun. 2016, 469, 222–228.
- Park, S.L.; Chung, T.-W.; Kim, S.; Hwang, B.; Kim, J.M.; Lee, H.M.; Cha, H.-J.; Seo, Y.; Choe, S.Y.; Ha, K.-T.; et al. HSP70-1 is required for interleukin-5-induced angiogenic responses through eNOS pathway. Sci. Rep. 2017, 7, 44687.
- Kluger, H.M.; Goel, H.L.; Breen, M.; Zhang, J.; Das, I.; Aznavoorian-Cheshire, S.; Greenberg, N.M.; Elgavish, A.; Languino, L. Using a Xenograft Model of Human Breast Cancer Metastasis to Find Genes Associated with Clinically Aggressive Disease. Cancer Res. 2005, 65, 5578–5587.
- Teng, Y.; Ngoka, L.; Mei, Y.; Lesoon, L.; Cowell, J. HSP90 and HSP70 Proteins Are Essential for Stabilization and Activation of WASF3 Metastasis-promoting Protein*. J. Biol. Chem. 2012, 287, 10051–10059.
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626.
- Kasioumi, P.; Vrazeli, P.; Vezyraki, P.; Zerikiotis, S.; Katsouras, C.; Damalas, A.; Angelidis, C. Hsp70 (HSP70A1A) downregulation enhances the metastatic ability of cancer cells. Int. J. Oncol. 2018, 54, 821–832.
- Mao, H.; Li, F.; Ruchalski, K.; Mosser, D.D.; Schwartz, J.; Wang, Y.; Borkan, S.C.; Stortchevoi, A.; Varshney, U.; Rajbhandary, U.L. hsp72 Inhibits Focal Adhesion Kinase Degradation in ATP-depleted Renal Epithelial Cells. J. Biol. Chem. 2003, 278, 18214–18220.
- Jang, K.W.; Lee, J.E.; Kim, S.Y.; Kang, M.-W.; Na, M.H.; Lee, C.S.; Song, K.S.; Lim, S.P. The C-terminus of Hsp70-Interacting Protein Promotes Met Receptor Degradation. J. Thorac. Oncol. 2011, 6, 679–687.
- Iii, J.K.; Jinwal, U.K.; Jin, Y.; O’Leary, J.; Jones, J.; Johnson, A.G.; Blair, L.J.; Abisambra, J.F.; Chang, L.; Miyata, Y.; et al. Facilitating Akt Clearance via Manipulation of Hsp70 Activity and Levels*. J. Biol. Chem. 2009, 285, 2498–2505.
- Li, H.; Li, Y.; Liu, D.; Sun, H.; Su, N.; Yang, F.; Liu, J. Extracellular HSP70/HSP70-PCs Promote Epithelial-Mesenchymal Transition of Hepatocarcinoma Cells. PLoS ONE 2013, 8, e84759.
- Komarova, E.; Marchenko, L.; Zhakhov, A.; Nikotina, A.; Aksenov, N.; Suezov, R.; Ischenko, A.; Margulis, B.; Guzhova, I.V. Extracellular Hsp70 Reduces the Pro-Tumor Capacity of Monocytes/Macrophages Co-Cultivated with Cancer Cells. Int. J. Mol. Sci. 2019, 21, 59.
- Mao, H.; Li, Z.; Zhou, Y.; Li, Z.; Zhuang, S.; An, X.; Zhang, B.; Chen, W.; Nie, J.; Wang, Z.; et al. HSP72 attenuates renal tubular cell apoptosis and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Physiol. 2008, 295, F202–F214.
- Yun, C.-H.; Yoon, S.-Y.; Nguyen, T.T.; Cho, H.-Y.; Kim, T.-H.; Kim, S.-T.; Kim, B.-C.; Hong, Y.-S.; Kim, S.-J.; Lee, H.J. Geldanamycin inhibits TGF-β signaling through induction of Hsp70. Arch. Biochem. Biophys. 2010, 495, 8–13.
- Li, Y.; Kang, X.; Wang, Q. HSP70 decreases receptor-dependent phosphorylation of Smad2 and blocks TGF-β-induced epithelial-mesenchymal transition. J. Genet. Genom. 2011, 38, 111–116.
- Liu, J.; Bao, J.; Hao, J.; Peng, Y.; Hong, F. HSP70 inhibits high glucose-induced Smad3 activation and attenuates epithelial-to-mesenchymal transition of peritoneal mesothelial cells. Mol. Med. Rep. 2014, 10, 1089–1095.
- Jeon, H.-M.; Lee, J. MET: Roles in epithelial-mesenchymal transition and cancer stemness. Ann. Transl. Med. 2017, 5, 5.
- Chao, A.; Lai, C.-H.; Tsai, C.-L.; Hsueh, S.; Hsueh, C.; Lin, C.-Y.; Chou, H.-H.; Lin, Y.-J.; Chen, H.-W.; Chang, T.-C.; et al. Tumor Stress-Induced Phosphoprotein1 (STIP1) as a Prognostic Biomarker in Ovarian Cancer. PLoS ONE 2013, 8, e57084.
- Da Fonseca, A.C.C.; Wang, H.; Fan, H.; Chen, X.; Zhang, I.; Zhang, L.; Lima, F.R.S.; Badie, B. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 2014, 274, 71–77.
- Willmer, T.; Contu, L.; Blatch, G.; Edkins, A.L. Knockdown of Hop downregulates RhoC expression, and decreases pseudopodia formation and migration in cancer cell lines. Cancer Lett. 2013, 328, 252–260.
- Walsh, N.; Larkin, A.; Swan, N.; Conlon, K.; Dowling, P.; McDermott, R.; Clynes, M. RNAi knockdown of Hop (Hsp70/Hsp90 organising protein) decreases invasion via MMP-2 down regulation. Cancer Lett. 2011, 306, 180–189.
- Suzuki, M.; Iwasaki, M.; Sugio, A.; Hishiya, A.; Tanaka, R.; Endo, T.; Takayama, S.; Saito, T. BAG3 (BCL2-associated athanogene 3) interacts with MMP-2 to positively regulate invasion by ovarian carcinoma cells. Cancer Lett. 2011, 303, 65–71.
- Lee, K.-J.; Kim, Y.M.; Kim, D.Y.; Jeoung, O.; Han, K.; Lee, S.-T.; Lee, Y.-S.; Park, K.H.; Park, J.H.; Kim, D.J.; et al. Release of heat shock protein 70 (Hsp70) and the effects of extracellular Hsp70 on matric metalloproteinase-9 expression in human monocytic U937 cells. Exp. Mol. Med. 2006, 38, 364–374.
- Chetty, C.; Vanamala, S.K.; Gondi, C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. MMP-9 induces CD44 cleavage and CD44 mediated cell migration in glioblastoma xenograft cells. Cell. Signal. 2011, 24, 549–559.
- Sossey-Alaoui, K.; Li, X.; Ranalli, T.A.; Cowell, J. WAVE3-mediated Cell Migration and Lamellipodia Formation Are Regulated Downstream of Phosphatidylinositol 3-Kinase*. J. Biol. Chem. 2005, 280, 21748–21755.
- Teng, Y.; Ren, M.Q.; Cheney, R.; Sharma, S.; Cowell, J. Inactivation of the WASF3 gene in prostate cancer cells leads to suppression of tumorigenicity and metastases. Br. J. Cancer 2010, 103, 1066–1075.
- Sossey-Alaoui, K.; Safina, A.; Li, X.; Vaughan, M.M.; Hicks, D.G.; Bakin, A.; Cowell, J. Down-Regulation of WAVE3, a Metastasis Promoter Gene, Inhibits Invasion and Metastasis of Breast Cancer Cells. Am. J. Pathol. 2007, 170, 2112–2121.
- Breuninger, S.; Stangl, S.; Werner, C.; Sievert, W.; Lobinger, D.; Foulds, G.; Wagner, S.; Pickhard, A.; Piontek, G.; Kokowski, K.; et al. Membrane Hsp70—A Novel Target for the Isolation of Circulating Tumor Cells After Epithelial-to-Mesenchymal Transition. Front. Oncol. 2018, 8, 497.
- Farkas, B.; Hantschel, M.; Magyarlaki, M.; Becker, B.; Scherer, K.; Landthaler, M.; Pfister, K.; Gehrmann, M.; Gross, C.; Mackensen, A.; et al. Heat shock protein 70 membrane expression and melanoma-associated marker phenotype in primary and metastatic melanoma. Melanoma Res. 2003, 13, 147–152.
- Botzler, C.; Schmidt, J.; Luz, A.; Jennen, L.; Issels, R.; Multhoff, G. Differential Hsp70 plasma-membrane expression on primary human tumors and metastases in mice with severe combined immunodeficiency. Int. J. Cancer 1998, 77, 942–948.
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155.
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA USA 2010, 108, 733–738.
- Williamson, D.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Foloppe, N.; Francis, G.L.; Graham, C.J.; Howes, R.; et al. Novel Adenosine-Derived Inhibitors of 70 kDa Heat Shock Protein, Discovered Through Structure-Based Design†. J. Med. Chem. 2009, 52, 1510–1513.
- Macias, A.; Williamson, D.; Allen, N.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Francis, G.L.; Graham, C.J.; et al. Adenosine-Derived Inhibitors of 78 kDa Glucose Regulated Protein (Grp78) ATPase: Insights into Isoform Selectivity. J. Med. Chem. 2011, 54, 4034–4041.
- Li, X.; Colvin, T.; Rauch, J.N.; Acosta-Alvear, D.; Kampmann, M.; Dunyak, B.; Hann, B.; Aftab, B.T.; Murnane, M.; Cho, M.; et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol. Cancer Ther. 2015, 14, 642–648.
- Yaglom, J.A.; Wang, Y.; Li, A.; Li, Z.; Monti, S.; Alexandrov, I.; Lu, X.; Sherman, M.Y. Cancer cell responses to Hsp70 inhibitor JG-98: Comparison with Hsp90 inhibitors and finding synergistic drug combinations. Sci. Rep. 2018, 8, 3010.
- Sherman, M.; Gabai, V. Hsp70 in cancer: Back to the future. Oncogene 2014, 34, 4153–4161.
- Koren, J.; Miyata, Y.; Kiray, J.; O’Leary, J.C.; Nguyen, L.; Guo, J.; Blair, L.J.; Li, X.; Jinwal, U.K.; Cheng, J.Q.; et al. Correction: Rhodacyanine Derivative Selectively Targets Cancer Cells and Overcomes Tamoxifen Resistance. PLoS ONE 2012, 7.
- Massey, A.J.; Williamson, D.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2009, 66, 535–545.
- Deocaris, C.C.; Widodo, N.; Shrestha, B.G.; Kaur, K.; Ohtaka, M.; Yamasaki, K.; Kaul, S.C.; Wadhwa, R. Mortalin sensitizes human cancer cells to MKT-077-induced senescence. Cancer Lett. 2007, 252, 259–269.
- Leu, J.I.-J.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A Small Molecule Inhibitor of Inducible Heat Shock Protein 70. Mol. Cell 2009, 36, 15–27.
- Schmitt, E.; Maingret, L.; Puig, P.-E.; Rérole, A.-L.; Hammann, A.; Ghiringhelli, F.; Solary, E.; Kroemer, G.; Garrido, C. Heat Shock Protein 70 Neutralization Exerts Potent Antitumor Effects in Animal Models of Colon Cancer and Melanoma. Cancer Res. 2006, 66, 4191–4197.
- Ernst, K.; Liebscher, M.; Mathea, S.; Granzhan, A.; Schmid, J.; Popoff, M.R.; Ihmels, H.; Barth, H.; Schiene-Fischer, C. A novel Hsp70 inhibitor prevents cell intoxication with the actin ADP-ribosylating Clostridium perfringens iota toxin. Sci. Rep. 2016, 6, 20301.
- Rérole, A.-L.; Gobbo, J.; De Thonel, A.; Schmitt, E.; De Barros, J.P.P.; Hammann, A.; Lanneau, D.; Fourmaux, E.; Demidov, O.; Micheau, O.; et al. Peptides and Aptamers Targeting HSP70: A Novel Approach for Anticancer Chemotherapy. Cancer Res. 2011, 71, 484–495.
- Rodina, A.; Patel, P.D.; Kang, Y.; Patel, Y.; Baaklini, I.; Wong, M.J.; Taldone, T.; Yan, P.; Yang, C.; Maharaj, R.; et al. Identification of an Allosteric Pocket on Human Hsp70 Reveals a Mode of Inhibition of This Therapeutically Important Protein. Chem. Biol. 2013, 20, 1469–1480.
- Williams, D.R.; Park, S.; Shin, I.; Ko, S.-K.; Lee, M.-R. An Apoptosis-Inducing Small Molecule That Binds to Heat Shock Protein 70. Angew. Chem. Int. Ed. 2008, 47, 7466–7469.
- Ko, S.-K.; Kim, J.; Na, D.C.; Park, S.; Park, S.-H.; Hyun, J.Y.; Baek, K.-H.; Kim, N.D.; Kim, N.-K.; Park, Y.N.; et al. A Small Molecule Inhibitor of ATPase Activity of HSP70 Induces Apoptosis and Has Antitumor Activities. Chem. Biol. 2015, 22, 391–403.
- Braunstein, M.J.; Scott, S.S.; Scott, C.M.; Behrman, S.; Walter, P.; Wipf, P.; Coplan, J.D.; Chrico, W.; Joseph, D.; Brodsky, J.L.; et al. Antimyeloma Effects of the Heat Shock Protein 70 Molecular Chaperone Inhibitor MAL3-101. J. Oncol. 2011, 2011, 1–11.
- Fewell, S.W.; Smith, C.M.; Lyon, M.A.; Dumitrescu, T.P.; Wipf, P.; Day, B.W.; Brodsky, J.L. Small Molecule Modulators of Endogenous and Co-chaperone-stimulated Hsp70 ATPase Activity. J. Biol. Chem. 2004, 279, 51131–51140.
- Huryn, D.; Brodsky, J.L.; Brummond, K.M.; Chambers, P.G.; Eyer, B.; Ireland, A.W.; Kawasumi, M.; Laporte, M.G.; Lloyd, K.; Manteau, B.; et al. Chemical methodology as a source of small-molecule checkpoint inhibitors and heat shock protein 70 (Hsp70) modulators. Proc. Natl. Acad. Sci. USA USA 2011, 108, 6757–6762.
- Phillips, P.; Sangwan, V.; Borja-Cacho, D.; Dudeja, V.; Vickers, S.; Saluja, A.K. Myricetin induces pancreatic cancer cell death via the induction of apoptosis and inhibition of the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Cancer Lett. 2011, 308, 181–188.
- Howe, M.K.; Bodoor, K.; Carlson, D.A.; Hughes, P.F.; Alwarawrah, Y.; Loiselle, D.R.; Jaeger, A.M.; Darr, D.B.; Jordan, J.L.; Hunter, L.M.; et al. Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem. Biol. 2014, 21, 1648–1659.
- Phillips, P.A.; Dudeja, V.; McCarroll, J.A.; Borja-Cacho, D.; Dawra, R.K.; Grizzle, W.E.; Vickers, S.M.; Saluja, A.K. Triptolide Induces Pancreatic Cancer Cell Death via Inhibition of Heat Shock Protein 70. Cancer Res. 2007, 67, 9407–9416.
- Banerjee, S.; Thayanithy, V.; Sangwan, V.; MacKenzie, T.N.; Saluja, A.K.; Subramanian, S. Minnelide reduces tumor burden in preclinical models of osteosarcoma. Cancer Lett. 2013, 335, 412–420.
- Antonoff, M.B.; Chugh, R.; Skube, S.J.; Dudeja, V.; Borja-Cacho, D.; Clawson, K.A.; Vickers, S.M.; Saluja, A.K. Role of Hsp-70 in Triptolide-Mediated Cell Death of Neuroblastoma. J. Surg. Res. 2010, 163, 72–78.
- MacKenzie, T.N.; Mujumdar, N.; Banerjee, S.; Sangwan, V.; Sarver, A.; Vickers, S.; Subramanian, S.; Saluja, A.K. Triptolide induces the expression of miR-142-3p: A negative regulator of heat shock protein 70 and pancreatic cancer cell proliferation. Mol. Cancer Ther. 2013, 12, 1266–1275.
- Jacobson, B.A.; Chen, E.Z.; Tang, S.; Belgum, H.S.; McCauley, J.A.; Evenson, K.A.; Etchison, R.G.; Jay-Dixon, J.; Patel, M.; Raza, A.; et al. Triptolide and its prodrug minnelide suppress Hsp70 and inhibit in vivo growth in a xenograft model of mesothelioma. Genes Cancer 2015, 6, 144–152.
- Rousalova, I.; Banerjee, S.; Sangwan, V.; Evenson, K.; McCauley, J.A.; Kratzke, R.; Vickers, S.M.; Saluja, A.K.; D’Cunha, J. Minnelide: A Novel Therapeutic That Promotes Apoptosis in Non-Small Cell Lung Carcinoma In Vivo. PLoS ONE 2013, 8, e77411.
- Liu, Q. Triptolide and its expanding multiple pharmacological functions. Int. Immunopharmacol. 2011, 11, 377–383.
- Propper, D.; Han, H.; Von Hoff, D.; Borazanci, E.; Reya, T.; Ghergurovich, J.; Pshenichnaya, I.; Antal, C.; Condjella, R.; Sharma, S.; et al. Abstract CT165: Phase II open label trial of minnelide™ in patients with chemotherapy refractory metastatic pancreatic cancer. Clin. Trials 2019.
- Hung, C.-M.; Su, Y.-H.; Lin, H.-Y.; Lin, J.-N.; Liu, L.-C.; Ho, C.-T.; Way, T.-D. Demethoxycurcumin Modulates Prostate Cancer Cell Proliferation via AMPK-Induced Down-regulation of HSP70 and EGFR. J. Agric. Food Chem. 2012, 60, 8427–8434.
- Jung, J.H.; Lee, J.O.; Kim, J.H.; Lee, S.K.; You, G.Y.; Park, S.H.; Park, J.M.; Kim, E.-K.; Suh, P.-G.; An, J.K.; et al. Quercetin suppresses HeLa cell viability via AMPK-induced HSP70 and EGFR down-regulation. J. Cell. Physiol. 2010, 223, 408–414.
- Choi, J.-A.; Kim, J.-Y.; Lee, J.-Y.; Kang, C.-M.; Kwon, H.-J.; Yoo, Y.D.; Kim, T.-W.; Lee, Y.-S.; Lee, S.-J. Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin. Int. J. Oncol. 2001, 19, 837–844.
- Milani, V.; Stangl, S.; Issels, R.; Gehrmann, M.; Wagner, B.; Hube, K.; Mayr, D.; Hiddemann, W.; Molls, M.; Multhoff, G. Anti-tumor activity of patient-derived NK cells after cell-based immunotherapy—A case report. J. Transl. Med. 2009, 7, 50.
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of Colon and Lung Cancer Patients with ex Vivo Heat Shock Protein 70-Peptide-Activated, Autologous Natural Killer Cells: A Clinical Phase I Trial. Clin. Cancer Res. 2004, 10, 3699–3707.
- Trimble, C.L.; Peng, S.; Kos, F.; Gravitt, P.; Viscidi, R.; Sugar, E.; Pardoll, E.; Wu, T. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin. Cancer Res. 2009, 15, 361–367.
- Maeda, Y.; Yoshimura, K.; Matsui, H.; Shindo, Y.; Tamesa, T.; Tokumitsu, Y.; Hashimoto, N.; Tokuhisa, Y.; Sakamoto, K.; Sakai, K.; et al. Dendritic cells transfected with heat-shock protein 70 messenger RNA for patients with hepatitis C virus-related hepatocellular carcinoma: A phase 1 dose escalation clinical trial. Cancer Immunol. Immunother. 2015, 64, 1047–1056.
- Kokowski, K.; Stangl, S.; Seier, S.; Hildebrandt, M.; Vaupel, P.; Multhoff, G. Radiochemotherapy combined with NK cell transfer followed by second-line PD-1 inhibition in a patient with NSCLC stage IIIb inducing long-term tumor control: A case study. Strahlenther. und Onkol. 2019, 195, 352–361.
- Li, Z.; Serrano, D.; Baglietto, L.; Johansson, H.; Mariette, F.; Torrisi, R.; Onetto, M.; Paganuzzi, M.; DeCensi, A. Combination of Imatinib Mesylate with Autologous Leukocyte-Derived Heat Shock Protein and Chronic Myelogenous Leukemia. Clin. Cancer Res. 2005, 11, 4460–4468.
- ClinicalTrials.gov. Available online: (accessed on 18 February 2020).
- Li, J.-L.; Liu, H.-L.; Zhang, X.-R.; Xu, J.-P.; Hu, W.-K.; Liang, M.; Chen, S.-Y.; Hu, F.; Chu, D.-T. A phase I trial of intratumoral administration of recombinant oncolytic adenovirus overexpressing HSP70 in advanced solid tumor patients. Gene Ther. 2008, 16, 376–382.

