 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | RUOJING ZHANG | + 6201 word(s) | 6201 | 2021-05-08 10:41:13 | | | |
| 2 | Conner Chen | Meta information modification | 6201 | 2021-05-25 06:08:30 | | |
Video Upload Options
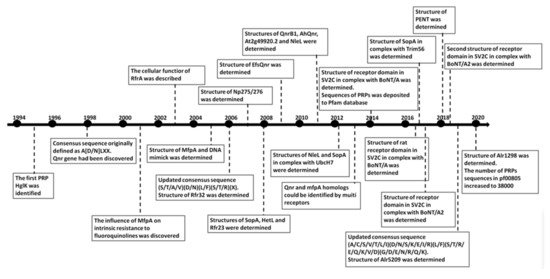
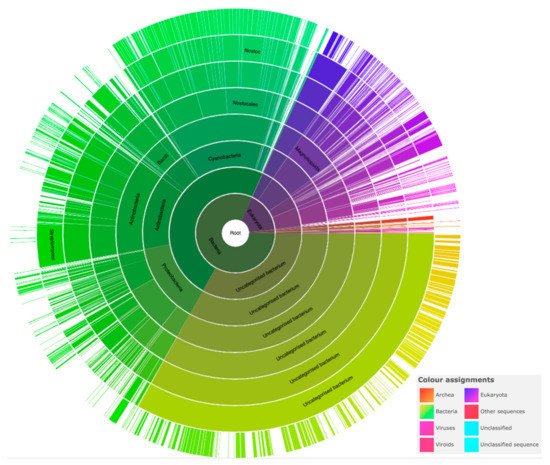
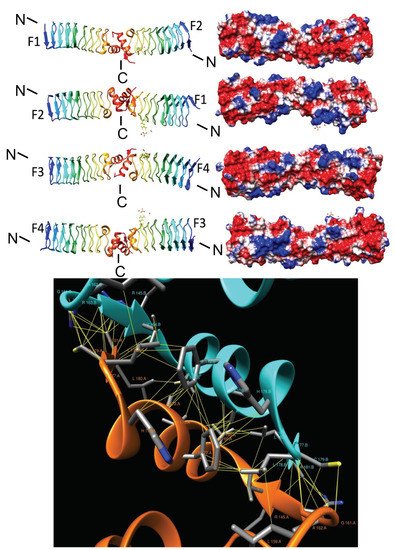
The pentapeptide repeat protein (PRP) superfamily, identified in 1998, has grown to nearly 39,000 sequences from over 3300 species. PRPs, recognized as having at least eight contiguous pentapeptide repeats (PRs) of a consensus pentapeptide sequence, adopt a remarkable structure, namely, a right-handed quadrilateral β-helix with four consecutive PRs forming a single β-helix coil. Adjacent coils join together to form a β-helix “tower” stabilized by β-ladders on the tower faces and type I, type II, or type IV β-turns facilitating an approximately −90° redirection of the polypeptide chain joining one coil face to the next. PRPs have been found in all branches of life, but they are predominantly found in cyanobacteria. Cyanobacteria have existed on earth for more than two billion years and are thought to be responsible for oxygenation of the earth’s atmosphere. Filamentous cyanobacteria such as Nostoc sp. strain PCC 7120 may also represent the oldest and simplest multicellular organisms known to undergo cell differentiation on earth.
1. Background
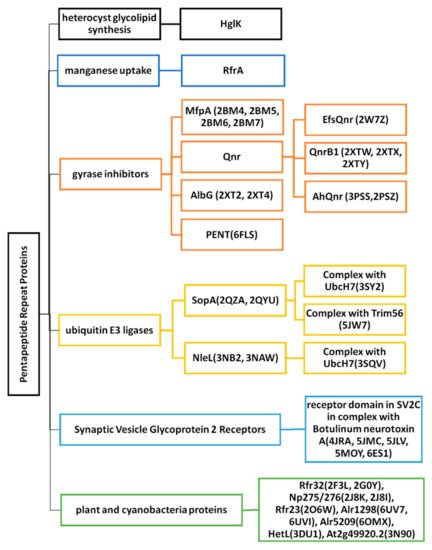
2. Cyanobacterial and Eubacterial PRPs with an Associated Biochemical or Cellular Function
2.1. Heterocyst Glycolipid Biosynthesis-HglK
2.2. Regulator of Manganese Uptake-RfrA
2.3. Gyrase Inhibitors
2.3.1. MfpA (2BM4, 2BM5, 2BM6 and 2BM7)
2.3.2. EfsQnr (2W7Z)
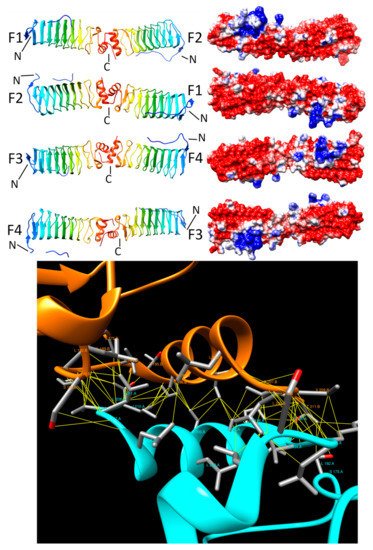
2.3.3. QnrB1 (2XTW, 2XTX, and 2XTY)
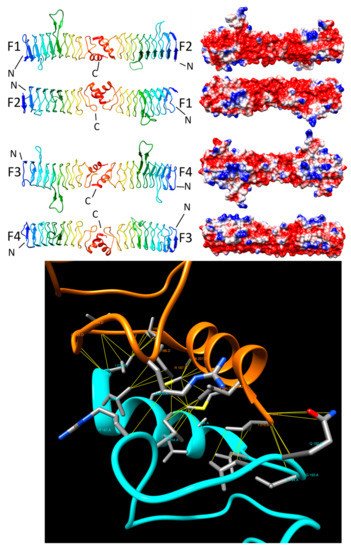
2.3.4. AhQnr (3PSS and 3PSZ)
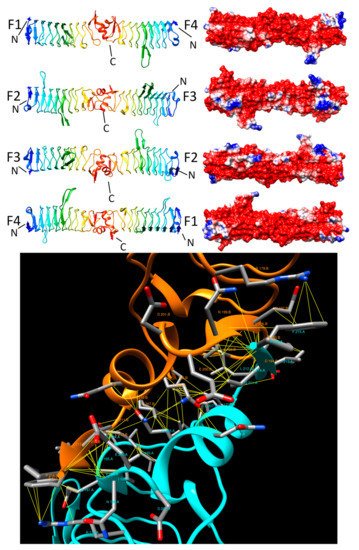
2.3.5. AlbG (2XT2 and 2XT4)
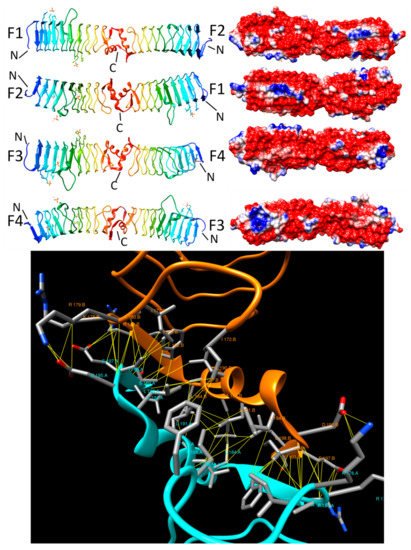
2.3.6. PENT (6FLS)
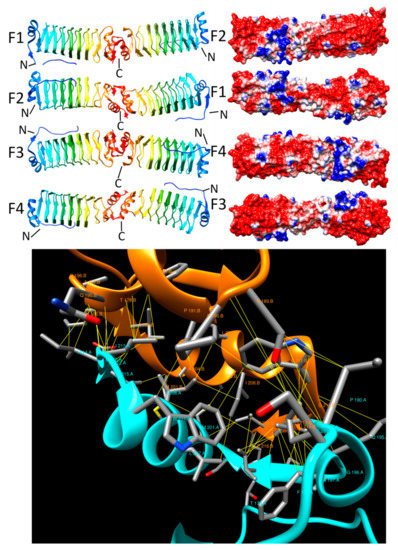
2.4. Ubiquitin E3 Ligases
2.4.1. SopA (2QYU, 2QZA, 3SY2 and 5JW7)
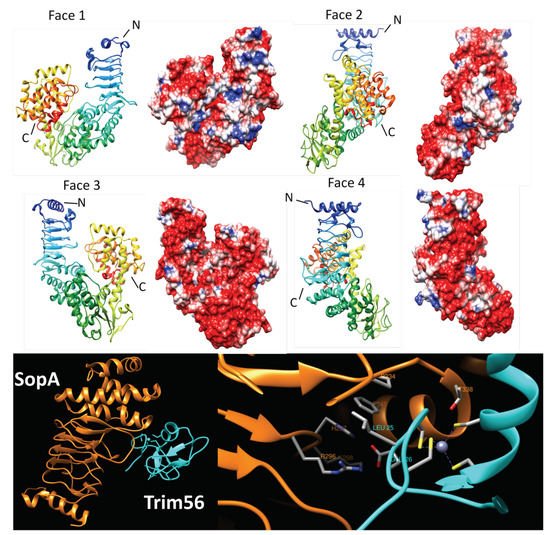
2.4.2. NleL (3NB2, 3NAW, and 3SQV)
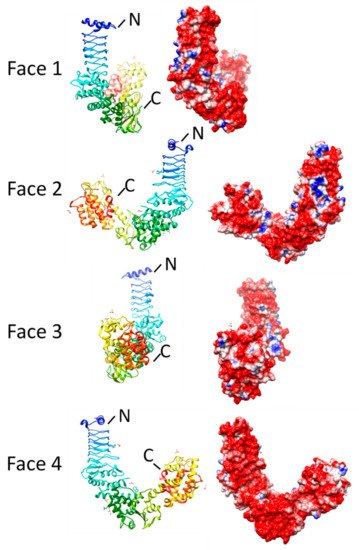
2.5. Synaptic Vesicle Glycoprotein 2 Receptors
SV2C-LD (4JRA, 5JMC, 5JLV, 5MOY, and 6ES1)
References
- Black, K.; Buikema, W.J.; Haselkorn, R. The hglK gene is required for localization of heterocyst-specific glycolipids in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 1995, 177, 6440–6448.
- Bateman, A.; Murzin, A.G.; Teichmann, S.A. Structure and distribution of pentapeptide repeats in bacteria. Protein Sci. 1998, 7, 1477–1480.
- Martínez-Martínez, L.; Pascual, A.; Jacoby, G.A. Quinolone resistance from a transferable plasmid. Lancet 1998, 351, 797–799.
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137.
- Montero, C.; Mateu, G.; Rodriguez, R.; Takiff, H. Intrinsic resistance of Mycobacterium smegmatis to fluoroquinolones may be influenced by new pentapeptide protein MfpA. Antimicrob. Agents Chemother. 2001, 45, 3387–3392.
- Hegde, S.S.; Vetting, M.W.; Roderick, S.L.; Mitchenall, L.A.; Maxwell, A.M.; Takiff, H.E.; Blanchard, J.S. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Abstr. Pap. Am. Chem. Soc. 2005, 230, U538–U539.
- Jacoby, G.A.; Hooper, D.C. Phylogenetic analysis of chromosomally determined qnr and related proteins. Antimicrob. Agents Chemother. 2013, 57, 1930–1934.
- Chandler, L.E.; Bartsevich, V.V.; Pakrasi, H.B. Regulation of Manganese Uptake in Synechocystis 6803 by RfrA, a Member of a Novel Family of Proteins Containing a Repeated Five-Residues Domain. Biochemistry 2003, 42, 5508–5514.
- Vetting, M.W.; Hegde, S.S.; Fajardo, J.E.; Fiser, A.; Roderick, S.L.; Takiff, H.E.; Mitchenall, L.A.; Maxwell, A. Pentapeptide repeat proteins. Biochemistry 2006, 5, 1–10.
- Buchko, G.W. Pentapeptide Repeat Proteins and Cyanobacteria. Handb. Cyanobacteria Biochem. Biotechnol. Appl. 2009, 233–257.
- Shah, S.; Heddle, J.G. Squaring up to DNA: Pentapeptide repeat proteins and DNA mimicry. Appl. Microbiol. Biotechnol. 2014, 98, 9545–9560.
- Ni, S.S.; Sheldrick, G.M.; Benning, M.M.; Kennedy, M.A. The 2 angstrom resolution crystal structure of HetL, a pentapeptide repeat protein involved in regulation of heterocyst differentiation in the cyanobacterium Nostoc sp. strain PCC 7120. J. Struct. Biol. 2009, 165, 47–52.
- Vetting, M.W.; Hegde, S.S.; Hazleton, K.Z.; Blanchard, J.S. Structural characterization of the fusion of two pentapeptide repeat proteins, Np275 and Np276, from Nostoc punctiforme: Resurrection of an ancestral protein. Protein Sci. 2007, 16, 755–760.
- Buchko, G.W.; Ni, S.S.; Robinson, H.; Welsh, E.A.; Pakrasi, H.B.; Kennedy, M.A. Characterization of two potentially universal turn motifs that shape the repeated five-residues fold-Crystal structure of a lumenal pentapeptide repeat protein from Cyanothece 51142. Protein Sci. 2006, 15, 2579–2595.
- Buchko, G.W.; Robinson, H.; Pakrasi, H.B.; Kennedy, M.A. Insights into the structural variation between pentapeptide repeat proteins-Crystal structure of Rfr23 from Cyanothece 51142. J. Struct. Biol. 2008, 162, 184–192.
- Ni, S.S.; McGookey, M.E.; Tinch, S.L.; Jones, A.N.; Jayaraman, S.; Tong, L.; Kennedy, M.A. The 1.7 A resolution structure of At2g44920, a pentapeptide-repeat protein in the thylakoid lumen of Arabidopsis thaliana. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2011, 67, 1480–1484.
- Vetting, M.W.; Hegde, S.S.; Blanchard, J.S. Crystallization of a pentapeptide-repeat protein by reductive cyclic pentylation of free amines with glutaraldehyde. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 462–469.
- Vetting, M.W.; Hegde, S.S.; Wang, M.H.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Structure of QnrB1, a Plasmid-mediated Fluoroquinolone Resistance Factor. J. Biol. Chem. 2011, 286, 25265–25273.
- Vetting, M.W.; Hegde, S.S.; Zhang, Y.; Blanchard, J.S. Pentapeptide-repeat proteins that act as topoisomerase poison resistance factors have a common dimer interface. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2011, 67, 296–302.
- Xiong, X.L.; Bromley, E.H.C.; Oelschlaeger, P.; Woolfson, D.N.; Spencer, J. Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: Conserved surface loops direct the activity of a Qnr protein from a Gram-negative bacterium. Nucleic Acids Res. 2011, 39, 3917–3927.
- Zhang, R.; Ni, S.; Kennedy, M.A. Type I beta turns make a new twist in pentapeptide repeat proteins: Crystal structure of Alr5209 from Nostoc sp. PCC 7120 determined at 1.7 angström resolution. J. Struct. Biol. X 2019, 3, 100010.
- Xu, S.; Kennedy, M.A. Structural dynamics of pentapeptide repeat proteins. Proteins 2020, 88, 1493–1512.
- Lechno-Yossef, S.; Fan, Q.; Wojciuch, E.; Wolk, C.P. Identification of Ten Anabaena sp. Genes That under Aerobic Conditions Are Required for Growth on Dinitrogen but Not for Growth on Fixed Nitrogen. J. Bacteriol. 2011, 193, 3482–3489.
- Buikema, W.J.; Haselkorn, R. Isolation and complementation of nitrogen fixation mutants of the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 1991, 173, 1879–1885.
- Arévalo, S.; Flores, E. Pentapeptide-repeat, cytoplasmic-membrane protein HglK influences the septal junctions in the heterocystous cyanobacterium Anabaena. Mol. Microbiol. 2020, 113, 794–806.
- Botello-Morte, L.; González, A.; Bes, M.T.; Peleato, M.L.; Fillat, M.F. Chapter Four-Functional Genomics of Metalloregulators in Cyanobacteria. In Advances in Botanical Research; Chauvat, F., Cassier-Chauvat, C., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 65, pp. 107–156.
- Vilchèze, C.; Jacobs, W.R., Jr. Resistance to Isoniazid and Ethionamide in Mycobacterium tuberculosis: Genes, Mutations, and Causalities. Microbiol. Spectr. 2014, 2, Mgm2-0014-2013.
- Ray, N.; Cavin, X.; Paul, J.C.; Maigret, B. Intersurf: Dynamic interface between proteins. J. Mol. Graph. Model. 2005, 23, 347–354.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612.
- Arsene, S.; Leclercq, R. Role of a qnr-like gene in the intrinsic resistance of Enterococcus faecalis to fluoroquinolones. Antimicrob. Agents Chemother. 2007, 51, 3254–3258.
- Tran, J.H.; Jacoby, G.A. Mechanism of plasmid-mediated quinolone resistance. Proc. Natl. Acad. Sci. USA 2002, 99, 5638–5642.
- Strahilevitz, J.; Jacoby, G.A.; Hooper, D.C.; Robicsek, A. Plasmid-mediated quinolone resistance: A multifaceted threat. Clin. Microbiol. Rev. 2009, 22, 664–689.
- Li, X.J.; Zhang, Y.J.; Zhou, X.T.; Hu, X.L.; Zhou, Y.X.; Liu, D.; Maxwell, A.; Mi, K.X. The plasmid-borne quinolone resistance protein QnrB, a novel DnaA-binding protein, increases the bacterial mutation rate by triggering DNA replication stress. Mol. Microbiol. 2019, 111, 1529–1543.
- Jacoby, G.A.; Walsh, K.E.; Mills, D.M.; Walker, V.J.; Oh, H.; Robicsek, A.; Hooper, D.C. qnrB, another plasmid-mediated gene for quinolone resistance. Antimicrob. Agents Chemother. 2006, 50, 1178–1182.
- Wang, M.H.; Guo, Q.L.; Xu, X.G.; Wang, X.Y.; Ye, X.Y.; Wu, S.; Hooper, D.C.; Wang, M.G. New Plasmid-Mediated Quinolone Resistance Gene, qnrC, Found in a Clinical Isolate of Proteus mirabilis. Antimicrob. Agents Chemother. 2009, 53, 1892–1897.
- Cavaco, L.M.; Frimodt-Moller, N.; Hasman, H.; Guardabassi, L.; Nielsen, L.; Aarestrup, F.M. Prevalence of quinolone resistance mechanisms and associations to minimum inhibitory concentrations in quinolone-resistant Escherichia coli isolated from humans and swine in Denmark. Microb. Drug Resist. 2008, 14, 163–169.
- Albornoz, E.; Tijet, N.; De Belder, D.; Gomez, S.; Martino, F.; Corso, A.; Melano, R.G.; Petroni, A. qnrE1, a Member of a New Family of Plasmid-Located Quinolone Resistance Genes, Originated from the Chromosome of Enterobacter Species. Antimicrob. Agents Chemother. 2017, 61, e02555-16.
- Hata, M.; Suzuki, M.; Matsumoto, M.; Takahashi, M.; Sato, K.; Ibe, S.; Sakae, K. Cloning of a novel gene for quinolone resistance from a transferable plasmid in Shigella flexneri 2b. Antimicrob. Agents Chemother. 2005, 49, 801–803.
- Fonseca, E.L.; Dos Santos Freitas, F.; Vieira, V.V.; Vicente, A.C. New qnr gene cassettes associated with superintegron repeats in Vibrio cholerae O1. Emerg. Infect Dis. 2008, 14, 1129–1131.
- Zhan, Y.Y.; Zheng, Z.W.; Chan, E.W.C.; Dong, N.; Xia, X.D.; Chen, S. Molecular Characterization of qnrVC Genes and Their Novel Alleles in Vibrio spp. Isolated from Food Products in China. Antimicrob. Agents Chemother. 2018, 62, e00529-18.
- Ruiz, J. Transferable Mechanisms of Quinolone Resistance from 1998 Onward. Clin. Microbiol. Rev. 2019, 32, e00007-19.
- Hashimi, S.M.; Wall, M.K.; Smith, A.B.; Maxwell, A.; Birch, R.G. The phytotoxin albicidin is a novel inhibitor of DNA gyrase. Antimicrob. Agents Chemother. 2007, 51, 181–187.
- Notari, L.; Martinez-Carranza, M.; Farias-Rico, J.A.; Stenmark, P.; von Heijne, G. Cotranslational Folding of a Pentarepeat beta-Helix Protein. J. Mol. Biol. 2018, 430, 5196–5206.
- Diao, J.; Zhang, Y.; Huibregtse, J.M.; Zhou, D.; Chen, J. Crystal structure of SopA, a Salmonella effector protein mimicking a eukaryotic ubiquitin ligase. Nat. Struct. Mol. Biol. 2008, 15, 65–70.
- Hayes, C.S.; Aoki, S.K.; Low, D.A. Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 2010, 44, 71–90.
- Galan, J.E.; Wolf-Watz, H. Protein delivery into eukaryotic cells by type III secretion machines. Nature 2006, 444, 567–573.
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 ubiquitin ligases—Emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133, jcs228072.
- Zhang, Y.; Higashide, W.M.; McCormick, B.A.; Chen, J.; Zhou, D.G. The inflammation-associated Salmonella SopA is a HECT-like E3 ubiquitin ligase. Mol. Microbiol. 2006, 62, 786–793.
- Fiskin, E.; Bhogaraju, S.; Herhaus, L.; Kalayil, S.; Hahn, M.; Dikic, I. Structural basis for the recognition and degradation of host TRIM proteins by Salmonella effector SopA. Nat. Commun. 2017, 8, 14004.
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434.
- Kamanova, J.; Sun, H.; Lara-Tejero, M.; Galan, J.E. The Salmonella Effector Protein SopA Modulates Innate Immune Responses by Targeting TRIM E3 Ligase Family Members. PLoS Pathog. 2016, 12, e1005552.
- Lin, D.Y.W.; Diao, J.B.; Zhou, D.G.; Chen, J. Biochemical and Structural Studies of a HECT-like Ubiquitin Ligase from Escherichia coli O157:H7. J. Biol. Chem. 2011, 286, 441–449.
- Sheng, X.; You, Q.; Zhu, H.; Chang, Z.; Li, Q.; Wang, H.; Wang, C.; Wang, H.; Hui, L.; Du, C.; et al. Bacterial effector NleL promotes enterohemorrhagic E. coli-induced attaching and effacing lesions by ubiquitylating and inactivating JNK. PLoS Pathog. 2017, 13, e1006534.
- Sheng, X.; You, Q.; Zhu, H.; Li, Q.; Gao, H.; Wang, H.; You, C.; Meng, Q.; Nie, Y.; Zhang, X.; et al. Enterohemorrhagic E. coli effector NleL disrupts host NF-kappaB signaling by targeting multiple host proteins. J. Mol. Cell Biol. 2020, 12, 318–321.
- Lin, D.Y.W.; Diao, J.B.; Chen, J. Crystal structures of two bacterial HECT-like E3 ligases in complex with a human E2 reveal atomic details of pathogen-host interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 1925–1930.
- Janz, R.; Goda, Y.; Geppert, M.; Missler, M.; Südhof, T.C. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 1999, 24, 1003–1016.
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 Is the Protein Receptor for Botulinum Neurotoxin A. Science 2006, 312, 592–596.
- Yao, G.R.; Zhang, S.C.; Mahrhold, S.; Lam, K.H.; Stern, D.; Bagramyan, K.; Perry, K.; Kalkum, M.; Rummel, A.; Dong, M.; et al. N-linked glycosylation of SV2 is required for binding and uptake of botulinum neurotoxin A. Nat. Struct. Mol. Biol. 2016, 23, 656–662.
- Benoit, R.M.; Frey, D.; Hilbert, M.; Kevenaar, J.T.; Wieser, M.M.; Stirnimann, C.U.; McMillan, D.; Ceska, T.; Lebon, F.; Jaussi, R.; et al. Structural basis for recognition of synaptic vesicle protein 2C by botulinum neurotoxin A. Nature 2014, 505, 108–111.
- Benoit, R.M.; Scharer, M.A.; Wieser, M.M.; Li, X.D.; Frey, D.; Kammerer, R.A. Crystal structure of the BoNT/A2 receptor-binding domain in complex with the luminal domain of its neuronal receptor SV2C. Sci. Rep. 2017, 7, 43588.
- Gustafsson, R.; Zhang, S.C.; Masuyer, G.; Dong, M.; Stenmark, P. Crystal Structure of Botulinum Neurotoxin A2 in Complex with the Human Protein Receptor SV2C Reveals Plasticity in Receptor Binding. Toxins 2018, 10, 153.


