Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alan Houghton | + 2284 word(s) | 2284 | 2021-05-17 11:20:09 | | | |
| 2 | Peter Tang | Meta information modification | 2284 | 2021-05-18 04:36:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Houghton, A. Natural Blues by Anthocyanins. Encyclopedia. Available online: https://encyclopedia.pub/entry/9725 (accessed on 27 July 2026).
Houghton A. Natural Blues by Anthocyanins. Encyclopedia. Available at: https://encyclopedia.pub/entry/9725. Accessed July 27, 2026.
Houghton, Alan. "Natural Blues by Anthocyanins" Encyclopedia, https://encyclopedia.pub/entry/9725 (accessed July 27, 2026).
Houghton, A. (2021, May 17). Natural Blues by Anthocyanins. In Encyclopedia. https://encyclopedia.pub/entry/9725
Houghton, Alan. "Natural Blues by Anthocyanins." Encyclopedia. Web. 17 May, 2021.
Copy Citation
Anthocyanins are a subclass of flavonoids; polyphenolic derivatives consisting of a fused benzopyrylium core (C- and A-rings) with an additional phenyl group attached at C2 (B-ring) formed by condensing a C6-C3 unit (p-coumaroyl-CoA) with three molecules of malonyl CoA. Although most anthocyanins are capable of producing blue colours in theory, at neutral pH (where the blue coloured anionic quinonoidal. base is formed) anthocyanins favour the formation of colourless species over the coloured species. The mechanisms whereby blue pigmentation by anthocyanins is achieved in nature are complex and multifold.
blue
anthocyanin
food colourants
co-pigment
quinonoid base
1. Introduction
Throughout history, our culture has been intrinsically linked to the colours of pigments and dyes accessible to us [1]. Colour is one of the primary visual indicators of food spoilage, so it comes as no surprise that we have evolved with innate psychological responses to abnormally coloured food; effects so strong that food not matching colour expectations can taste less appealing than otherwise identical, “colour-correct” foods [2]. With recent trends towards use of natural pigments in food, manufacturers have invested heavily in developing replacements for synthetic colourants. Despite plenty of options for yellow to purple, natural blue colourants remain elusive [3].
Currently, only three synthetic blue pigments are approved in Europe as food additives. Brilliant blue FCF (derived from petrochemicals), Indigo carmine (synthetic derivative of indigo), and patent blue V. However, only one natural pigment is available—phycocyanin, derived from “Spirulina”, which is composed of a mixture of three species of cyanobacteria, namely, Arthrospira platensis, Arthrospira fusiformis, and Arthrospira maxima.
Natural blue pigments are rare in nature, and only one known animal, a species of butterfly, Nessaea obrinus, can synthesise them. Instead, many apparently blue organisms create multi-layered nanostructures to reflect shorter wavelength light selectively, giving them their iridescent blue hues [4]. Unlike pigments however, the structural colour will change in these cases, depending on the viewing or illumination angle.
Plants produce an array of natural colourants, which can be broadly separated into four major groups: chlorophyll (green), carotenoids (yellow-red), betalains (yellow and red), and flavonoids (yellow-blue). Plants utilise these pigments to perform essential functions such as photosynthesis, radical scavenging, and attracting pollinators. Many fruits and vegetables are coloured with red and orange pigments, which have the greatest contrast against green foliage. This increases the likelihood they will be seen and eaten, allowing their seeds to be dispersed widely. This seed dispersal strategy is so effective that three unique red pigments have evolved: lycopene (a carotenoid found in fruits such as tomatoes), betacyanins (betalains found in some of the Caryophyllales) [5], and anthocyanins (such as the major pigment in strawberries, pelargonidin 3-O-glucoside) [6].
Despite the wide variety of pigments synthesised by plants, few blossoms or fruits are blue. In fact, some estimates suggest only 10% of angiosperm species include blue varieties. A major difficulty in producing blue pigments arises, in part, due to how pigments are coloured in the first place. Electrons in the highest occupied molecular orbital of chromophores (HOMO) can absorb a discrete amount of energy (corresponding to a specific wavelength of light) which promotes an electron to the lowest unoccupied molecular orbital (LUMO) [7]. The light reflected is a mixture of all unabsorbed wavelengths, which creates the colours we see. Alternating π bonds increase the delocalisation of electrons, reducing the energy an electron requires to move from the ground state to the excited state [8]. As the HOMO-LUMO energy gap shrinks, longer wavelength (lower energy) light is absorbed, causing a bluing in colour. To produce true blues, the absorbed wavelength of light is relatively long at around 580–620 nm.
Compounds with the conjugated systems necessary to absorb light in this range are typically large and complex, requiring multiple biosynthetic steps and heavy energy investment in their biosynthesis. Compounding this problem, blue colouration provides only a relatively niche evolutionary advantage in attracting specific pollinators [9]. Combined, these are the principal reasons that blue pigments are relatively uncommon in nature. However, blue-flowered varieties have been heavily selected by breeders and horticulturalists, increasing the range of blue flowers available to enthusiastic gardeners.
Synthetic colourants have been used historically to colour or colour-correct within the food industry. The safety of these colourants has rightfully been questioned, starting with the Food and Drug Act of 1906 in the USA, and more recently promoting a shift towards natural sources of pigment. The replacement of all colours with natural alternatives has proved challenging, particularly for blue pigments, due to their increased sensitivity to light, temperature, and oxygen, as well as their low water solubility compared to synthetic dyes.
However, a diverse array of stunning natural blue shades are available, particularly in floral systems, based on pigmentation by anthocyanins. Typically, anthocyanins are unstable in solution, especially under conditions where blue colouration could potentially be observed. This is due to the rapid formation of colourless forms, and the eventual isomerisation into yellow forms, leading to browning of products. To stabilise the anthocyanin quinonoid base and generate blue colours, anthocyanins form complexes through intramolecular co-pigmentation, intermolecular co-pigmentation, and metal chelation. Many of these strategies have been adopted in nature by plants that produce blue flowers or fruit.
2. General Features of Anthocyanins
Anthocyanins are a subclass of flavonoids; polyphenolic derivatives consisting of a fused benzopyrylium core (C- and A-rings) with an additional phenyl group attached at C2 (B-ring) formed by condensing a C6-C3 unit (p-coumaroyl-CoA) with three molecules of malonyl CoA (Table 1).
Table 1. General structures of common anthocyanins.
|
|
||
|---|---|---|
|
Anthocyanidin |
R1 |
R2 |
|
Pelargonidin |
H |
H |
|
Cyanidin |
H |
OH |
|
Delphinidin |
OH |
OH |
|
Peonidin |
OCH3 |
H |
|
Petunidin |
OH |
OCH3 |
|
Malvidin |
OCH3 |
OCH3 |
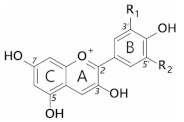
Twenty-seven naturally occurring anthocyanidins have been identified to date, however, six account for approximately 92% of all anthocyanins reported [10]. These six are pelargonidin, cyanidin, delphinidin, peonidin, petunidin, and malvidin (Table 1). The distributions of these six anthocyanidin classes in fruits and vegetables are cyanidin 50%, delphinidin 12%, pelargonidin 12%, peonidin 12%, petunidin 7%, and malvidin 7% [11].
Anthocyanidins are really only stable at very acid pH values (around pH 1); at physiological pH, they are quickly hydrated to form colourless forms (hemiketals). They are glycosylated at C3 in order to stabilise natural anthocyanidins, and these compounds are termed anthocyanins. Further decoration of the anthocyanin with carbohydrate, aromatic, and aliphatic moieties produces the wide array of anthocyanins seen in nature. It is these compounds which are responsible for almost all the orange/blue colours of floral systems.
Substitution of the B-ring with either hydroxyl or methoxyl groups influences the absorbance maxima of the anthocyanidin, and hence alters their colour from orange (pelargonidin) to bluish-purple (malvidin). Blue flowers commonly contain delphinidin derivatives, although there are a few exceptions where cyanidin-based anthocyanins confer blue [12].
The addition of hydroxyl groups on the B-ring tends to increase the maximum absorption of the anthocyanins in the visible range (λmax in nm) under specified conditions. Thus, for pelargonidin 3-glucose, cyanidin 3-glucoside, and delphinidin 3-glucoside, the respective visible λ values are 510, 530, and 543 nm in 1% HCl in methanol [13].
It is thought that due to the electronegativity of the oxygen atom in the B-ring, substituent groups would pull electron density away from the aromatic centre. The additional substituent groups in the ortho and para positions of the B-ring have a lone pair of electrons present on the oxygen atom and are adjacent to the sp2 orbital of the B-ring. Electrons are donated through resonance with the C2-C1’ bond, which becomes more electron-rich in the presence of ortho and para hydroxyl groups on the B-ring [14]. This increased electron density increases the energy of both the ground state (HOMO) and excited states (LUMO). As these interactions occur through the non-bonding orbital (which is energetically closer to the HOMO), the ground state experiences a greater magnitude of effect, effectively shrinking the transition energy gap and promoting absorption of longer wave-length light and a bathochromic shift.
3. Anthocyanin Multistate Equilibrium
All features of anthocyanin pigmentation need to be explained in the context of the multistate equilibrium which exists for anthocyanins in solution. Anthocyanins function as weak diacids due to their chromophore containing a strongly electron-withdrawing pyrilium ring, giving the phenolic hydroxyl groups at C4’, C5, and C7 fairly acidic properties. Of these hydroxyl groups, the C7-OH is the most acidic (pKa1 ≈ 4), followed by the C4’ hydroxyl (pKa2 > 7) for simple anthocyanins [15]. As weak diacids, anthocyanins become deprotonated as pH increases which, in turn, increases electron polarisation and increases the absorption maxima towards higher wavelengths.
Anthocyanin colour therefore is the result of the ratio of the three coloured species in equilibrium at any specific pH. At very acidic pH values (around pH < 2), the formation of the red flavylium ion (AH+) is favoured (Figure 1). This species is fully protonated and has a delocalised positive charge across the chromophore. As the pH increases above pKa1, the first deprotonation occurs, converting flavylium ions into the neutral quinonoid base (A) and the colour changes from red to purple. At pH values above pKa2, the quinonoid base is deprotonated further, forming the anionic quinonoid base (A−) with a negative delocalised charge [16] (Figure 1). This deprotonation sequence is accompanied by two shifts in λmax, the first (AH+→ A) typically of 20–30 nm and the second (A→ A−) a further shift of 50–60 nm [15].
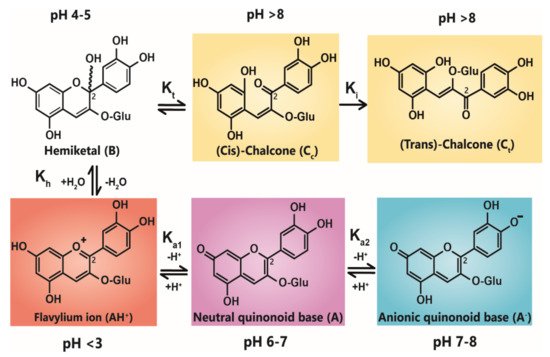
Figure 1. Structural transformation of cyanidin-3-O-glycoside as a function of pH. The three cyanidin species are given with their expected colours as outlined. Deprotonation from the C5-OH has been omitted for simplicity.
Due to the instability of A (and A−), for simple anthocyanins, the conversion of AH+ to A is generally less thermodynamically favourable than hydration at C2, giving the colourless hemiketal (B), and tautomerisation of B to give colourless isomeric chalcones (Ct and Cc). As the most stable products above pH 3, species B and chalcones deplete the available pool of the coloured species, and the red or purple colours begin to disappear as the pH is increased.
A beautiful example of how vacuolar pH can impact flower colour has been described for Japanese morning glory, which presents reddish brown flower buds that change to a brilliant blue as the petals unfold. This colour change accompanies an increase in pH of the vacuoles from 6.6 in buds to an unusually high value of 7.7 in mature petals. The increase in vacuolar pH is conferred by a tonoplast-located Na+/H+ exchanger [17][18], responsible for increasing the vacuolar pH by exchanging H+ for Na+ in vacuoles of petal cells during flower opening.
4. Co-Pigmentation
The diversity of colours observed in floral systems cannot be rationalised by differences in chemical substituent groups and the pH equilibrium alone; without interactions that stabilise the chromophore at physiological pH within the vacuole (which is usually about pH 5.5), most flowers would be devoid of their attractive hues and would instead be colourless or pale yellow (as a result of hemiketal and chalcone formation, respectively; Figure 1). Anthocyanins are large, planar compounds which lend themselves to the formation of non-covalent interactions with molecules termed co-pigments (Figure 2). Generally, these interactions are favourable towards the stabilisation of the coloured species through blocking hydration of the flavylium cation to the colourless hemiketal by the addition of water at C2 [19]. These interactions occur with the highest affinity between the coloured forms of the anthocyanin species and the co-pigment, and less so with the hemiketal and chalcone forms due to decreased conjugation of the tricyclic core upon ring opening [15] (Figure 2). Observations from the anthocyanins found typically in red wines have demonstrated these co-pigmentation interactions occur most strongly at around pH 3.6, or when A is the dominant species [20].
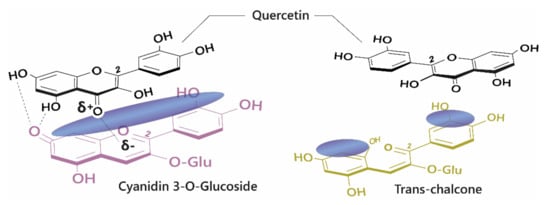
Figure 2. Left: Co-pigmentation interaction between cyanidin 3-O-glucoside and quercetin. Blue clouds above the structures denote polarisable surface area. Direct electrostatic interactions form between substituent groups, stabilising the coloured species. Right: Ring opening breaks the conjugated system across the tricyclic ring, reducing the planar surface for co-pigmentation.
5. Forces Involved in Co-Pigmentation
Hunter and Sanders [21] originally proposed that the π electron density of an unsubstituted aromatic ring results in a quadrupole moment, with a partial negative charge above both aromatic faces, and partial positive charge around the periphery. π-interaction is an attractive force between electrical quadrupoles, overpowering the repulsion of π-electron clouds. Where aromatic systems share a similar electron density, σ–π interactions are favourable and should therefore favour an edge-to-face or off-centre parallel arrangement, disfavouring face-centred arrangements (Figure 3).
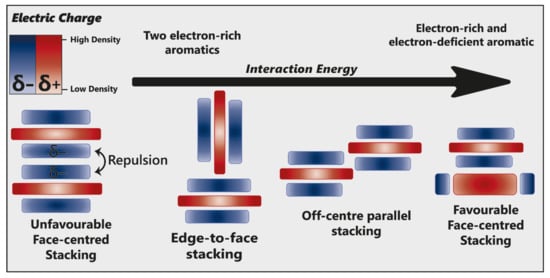
Figure 3. Alternative modes of stacking are shown, emphasising the locations of electrostatic attraction or repulsion. As the interaction energy increases, so too does complex stability. Adapted from [22].
The addition of electron-withdrawing substituents to an aromatic (arene) polarises π electron density away from the aromatic centre, reversing the direction of the quadrupole [22]. Pairs of electron-rich and electron-deficient aromatics preferentially pair in a face-centred arrangement, referred to as “aromatic donor–acceptor interaction” (Figure 3).
Pigment–co-pigment interactions are often attributed overwhelmingly to π–π interactions. However, these fail to account for other molecular forces which can initiate and drive co-pigmentation. The magnitude of the interaction is the product of all forces acting between the two aromatic systems, maximising electrostatic forces and dispersive attraction.
π–π interactions are not limited to arene moieties; cyclical aliphatic molecules have also been demonstrated to take part in these interactions, suggesting that the requirement for electron delocalisation (as seen in aromatics) is not strict [23]. This is particularly noteworthy within non-aromatic heterocycles, such as glucose, where the presence of an oxygen atom introduces asymmetry within the heterocycle, resulting in polarisation. Within any anthocyanin or co-pigment, there will be regions of high- and low-electron density. Therefore, the overall strength of intra- and intermolecular co-pigmentation interactions will depend on the alignment of these regions [21].
Such interactions may also explain why the addition a single glucose moiety at C3’ and C5’ (to produce ternatin C5) was sufficient to generate blue colouration in butterfly pea and also in genetically modified chrysanthemum [24][25]. These glucose groups may increase the dispersive attraction, and possibly direct electrostatic interactions, pulling the co-pigment closer to the chromophore and reducing the transition energy gap, thus producing a bluer colour.
References
- Barnett, J.R.; Miller, S.; Pearce, E. Colour and Art: A Brief History of Pigments. Opt. Laser Technol. 2006, 38, 445–453.
- Spence, C. On the Psychological Impact of Food Colour. Flavour 2015, 4, 21.
- Galaffu, N.; Bortlik, K.; Michel, M. An industry perspective on natural food colour stability. In Colour Additives for Foods and Beverages; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; pp. 91–130. ISBN 9781782420200.
- Moyroud, E.; Wenzel, T.; Middleton, R.; Rudall, P.J.; Banks, H.; Reed, A.; Mellers, G.; Killoran, P.; Westwood, M.M.; Steiner, U.; et al. Disorder in Convergent Floral Nanostructures Enhances Signalling to Bees. Nature 2017, 550, 469–474.
- Chandran, J.; Nisha, P.; Singhal, R.S.; Pandit, A.B. Degradation of Colour in Beetroot (Beta Vulgaris L.): A Kinetics Study. J. Food Sci. Technol. 2014, 51, 2678–2684.
- Lin-Wang, K.; McGhie, T.K.; Wang, M.; Liu, Y.; Warren, B.; Storey, R.; Espley, R.V.; Allan, A.C. Engineering the Anthocyanin Regulatory Complex of Strawberry (Fragaria Vesca). Front. Plant Sci. 2014, 5, 651.
- Englman, R.; Jortner, J. The Energy Gap Law for Radiationless Transitions in Large Molecules. Mol. Phys. 1970, 18, 285–287.
- Meier, H.; Stalmach, U.; Kolshorn, H. Effective Conjugation Length and UV/Vis Spectra of Oligomers. Acta Polym. 1997, 48, 379–384.
- Real, L.A. Uncertainty and Pollinator-Plant Interactions: The Foraging Behavior of Bees and Wasps on Artificial Flowers. Ecology 1981, 62, 20–26.
- Andersen, Ø.M.; Jordheim, M. Anthocyanins in health and disease - basic anthocyanin chemistry and dietary sources. In Anthocyanins in Health and Disease - Basic Anthocyanin Chemistry and Dietary Sources; Wallace, T.C., Giusti, M.M., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 30–107.
- Castañeda-Ovando, A.; de Pacheco-Hernández, M.L.; Páez-Hernández, M.E.; Rodríguez, J.A.; Galán-Vidal, C.A. Chemical Studies of Anthocyanins: A Review. Food Chem. 2009, 113, 859–871.
- Sasaki, N.; Nakayama, T. Achievements and Perspectives in Biochemistry Concerning Anthocyanin Modification for Blue Flower Coloration. Plant Cell Physiol. 2015, 56, 28–40.
- De Santiago, M.C.P.A.; Gouvêa, A.C.M.S.; de Godoy, R.L.O.; Borguini, R.G.; Pacheco, S.; Nogueira, R.I.; Nascimento, L.D.S.D.M.D.; Freitas, S.P. Analytical Standards Production for the Analysis of Pomegranate Anthocyanins by HPLC. Brazi. J. Food Technol. 2014, 17, 51–57.
- Mao, Y.; Head-Gordon, M.; Shao, Y. Unraveling Substituent Effects on Frontier Orbitals of Conjugated Molecules Using an Absolutely Localized Molecular Orbital Based Analysis. Chem. Sci. 2018, 9, 8598–8607.
- Dangles, O.; Fenger, J.-A. The Chemical Reactivity of Anthocyanins and Its Consequences in Food Science and Nutrition. Molecules 2018, 23, 1970.
- Basílio, N.; Pina, F. Chemistry and Photochemistry of Anthocyanins and Related Compounds: A Thermodynamic and Kinetic Approach. Molecules 2016, 21, 1502.
- Fukada-Tanaka, S.; Inagaki, Y.; Yamaguchi, T.; Saito, N.; Iida, S. Colour-Enhancing Protein in Blue Petals. Nature 2000, 407, 581.
- Yoshida, K.; Kondo, T.; Okazaki, Y.; Katou, K. Cause of Blue Petal Colour. Nature 1995, 373, 291.
- He, J.; Li, X.; Silva, G.T.M.; Quina, F.H.; Aquino, A.J.A. Quantum Chemical Investigation of the Intramolecular Copigmentation Complex of an Acylated Anthocyanin. Artic. J. Braz. Chem. Soc. 2019, 30, 492–498.
- Brouillard, R.; Wigand, M.C.; Dangles, O.; Cheminat, A. PH and Solvent Effects on the Copigmentation Reaction of Malvin with Polyphenols, Purine and Pyrimidine Derivatives. J. Chem. Soc. Perkin Trans. 1991, 2, 1235–1241.
- Hunter, C.A.; Sanders, J.K.M. The Nature of π-π Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534.
- Li, Y.; Prejanò, M.; Toscano, M.; Russo, N. Oenin and Quercetin Copigmentation: Highlights From Density Functional Theory. Front. Chem. 2018, 6, 245.
- Molcanov, K.; Kojić-Prodić, B. Towards Understanding π-Stacking Interactions between Non-Aromatic Rings. IUCrJ 2019, 6, 156–166.
- Marković, J.M.D.; Petranović, N.A.; Baranac, J.M. The Copigmentation Effect of Sinapic Acid on Malvin: A Spectroscopic Investigation on Colour Enhancement. J. Photochem. Photobiol. B Biol. 2005, 78, 223–228.
- Noda, N.; Yoshioka, S.; Kishimoto, S.; Nakayama, M.; Douzono, M.; Tanaka, Y.; Aida, R. Generation of Blue Chrysanthemums by Anthocyanin B-Ring Hydroxylation and Glucosylation and Its Coloration Mechanism. Sci. Adv. 2017, 3, e1602785.
More
Information
Subjects:
Plant Sciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.9K
Revisions:
2 times
(View History)
Update Date:
18 May 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No