 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Franziska Hornung | + 4191 word(s) | 4191 | 2021-05-08 09:02:08 | | | |
| 2 | Peter Tang | Meta information modification | 4191 | 2021-05-18 04:30:42 | | |
Video Upload Options
The alterations of adipocyte-derived signal mediators strongly influence the regulation of inflammation, resulting in chronic low-grade inflammation.
1. Introduction
Obesity is becoming a growing health problem worldwide. It is generally defined as a condition of increased adipose tissue mass [1] and can be further specified as an accumulation of body mass beyond physical requirements [2]. The WHO describes obesity and overweight as excessive fat accumulation leading to higher morbidity rates for various health problems [3]. In 1842, Adolphe Quetelet conducted pioneering work in analyzing the differences in the weight status of individuals [4]. The Quetelet dindex is calculated by dividing weight by height squared and is known nowadays as the Body Mass Index (BMI), which is still the most common way of classifying obesity [5]. Using this scale, overweight is defined as a BMI greater than or equal to 25 and obesity as a BMI greater than or equal to 30 [3].
With more than 1.9 billion overweight adults in 2016 and a worldwide tripling of the number of obese persons since 1975 [3], obesity has spread around the world and now affects a considerable part of the human population. This high prevalence represents a huge problem for our health care systems, because “Corpulence is not only a disease itself but the harbinger of others”, as Hippocrates already knew more than two millennia ago [6]. Metabolic diseases (e.g., type 2 diabetes mellitus), bone and soft tissue pathologies (e.g., osteoarthritis), and cardiovascular diseases (e.g., hypertension) belong to the main comorbidities of obesity. Moreover, obesity also leads to impaired lung function, increased occurrence of asthma, and obstructive sleep apnoea syndrome (OSA) [7]. Since obesity contributes to a wide variety of comorbidities, an excess of adipose tissue mass is expected to lead to a variety of molecular changes in the body. Regarding respiratory infections, an increased sensitivity has been observed, which might be connected to the above-mentioned increased incidence of comorbidities but also by a chronic low-grade inflammatory status.
In the process of infection progression, obese patients have been reported to show benefits or disadvantages compared to normal-weight subjects depending on the type of infection. On the one hand, the “obesity paradox” describes a benefit for the obese, e.g., in sepsis [8]. On the other hand, certain infections take a markedly more severe course in obese patients compared to normal-weight patients, as the current coronavirus disease (COVID-19) pandemic clearly illustrates [9][10]. Thus, the “obesity paradox” proposes that obese patients, although presenting numerous comorbidities, show a survival benefit [11].
2. The Inflammatory Activity of Adipose Tissue
Adipose tissue is classified into two main types, white adipose tissue (WAT) and brown adipose tissue (BAT). WAT is the more predominant form in the human body and plays a major role in energy storage. Thermogenesis is the main function of BAT. It was thought to be rather present in small mammals and human neonates [12]. Nedergaard et al. indeed revealed its presence in adult tissue [13].
Adipocytes are in general the main cell type within adipose tissue. They can be further classified according to their microscopical appearance [14]. A unilocular positioned lipid-vacuole characterizes white adipocytes, the predominant form in the WAT. Brown appearing adipocytes are defined by multilocular lipid-vacuoles and an increased amount of mitochondria, which is connected to their function in heat production in BAT. Both, WAT and BAT, are capable to transdifferentiate between the subtypes in response to the physiological conditions [15]. The intermediate cell forms are called beige adipocytes [16]. White adipocytes are additionally able to transdifferentiate into epithelial, milk-producing cells in the breast of pregnant women. These cells are named after their visual appearance, the pink adipocytes [17].
Besides adipocytes, also pre-adipocytes, endothelial cells, fibroblasts, leukocytes, and bone-marrow-derived macrophages are part of adipose tissue [18]. The number of macrophages positively correlates with body mass, adipocyte size, and expression of pro-inflammatory cytokines [19].
One of the main physiological functions of WAT is the regulation of fat reservoirs in the body via triacylglycerols (TG) stored in adipocytes. The mobilization and storage of TG must be well balanced with and connected to the energy intake and expenditure of the whole body. In this context, the lipolysis, i.e., the metabolization of TG, is also regulated by the autonomic nervous system, in particular adrenergic and cholinergic neurons [20].
In addition to energy storage, adipose tissue has an important endocrine function secreting a number of crucial soluble factors: Specific to adipose tissue are the so-called “adipocytokines” or “adipokines”, including, e.g., adiponectin, leptin, resistin, and visfatin [18]; described in more details in the next chapter. Other important produced factors include the cytokines tumor necrosis factor (TNF), interleukin-6 (IL-6), interleukin-1 (IL-1), CC-chemokine ligand 2 (CCL2), plasminogen activator inhibitor type I (PAI-I), and a number of complement factors [21][22]. Most of these factors are known as pro-inflammatory mediators that induce immune cell infiltration and play a major role in the development of infectious diseases.
According to the location of the deposition, adipose tissue can be further classified in subcutaneous adipose tissue (SCAT) and visceral adipose tissue (VAT). The excessive production of fat tissue during weight gain leads to a depletion of storage capacities and can result in ectopic lipid accumulation in visceral body cavities, skeletal muscle, or liver tissue [23][24]. This phenomenon can for instance play a major role in the development of insulin resistance [25].
An increase in adipose tissue mass during weight gain can be on a cellular level either orchestrated by an increase in the size of the adipocytes (hypertrophy) or their number (hyperplasia). Besides simple mass expansion, the tissue additionally passes through a process of remodeling characterized by extracellular matrix (ECM) overproduction, increased immune cell infiltration, and higher pro-inflammatory response [26].
Specifically, the crosstalk between adipocytes and macrophages plays an important role in the remodeling [27]. The elevated infiltration leads to the expression of macrophage-related inflammatory genes [28]. In this context, Sun et al. [26] proposed four different mechanisms as potential key initiator processes of macrophage migration to the adipose tissue: adipocyte death, chemotactic regulation, hypoxia, and fatty acid flux. Thus, the excess of body weight via increased adipose tissue mass results in a state of low-grade chronic inflammation (Figure 1).
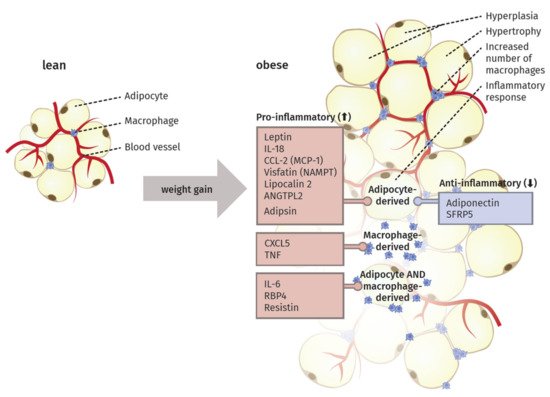
Saltiel et al. [29] suggested the occurrence of a “phenotypic switch” during polarization of macrophages from an anti-inflammatory M2 type to the M1 form. In lean individuals, the polarized M2 type seems to encourage the normal function of adipocytes by promoting repair of tissue and angiogenesis, i.e., the sprouting of new vessels to ensure an appropriate oxygen supply. In obese individuals, the M1 type is fostering an inflammatory milieu in the adipose tissue by secreting factors like TNF-α, inducible nitric oxide synthase (iNOS), C-C chemokine receptor type 2 (CCR2) or monocyte chemotactic protein 1 (MCP1). Sun et al. [27] proposed that this switch in polarized macrophages defines the fate of adipocyte function and the overall inflammatory profile of the tissue. They also discriminated between healthy and pathological adipose tissue expansion. In the former, an increase in mass happens mainly via a growing number of small adipocytes, recruitment of other stromal cells, maintenance of oxygen supply, and only minimal induction of ECM production and inflammation. In contrast, pathological expansion is defined as a rapid increase in the size of existing adipocytes with hypoxia due to decreased blood vessel formation, massive ECM deposition, and a higher number of macrophages, especially the pro-inflammatory M1 type, leading to a state of chronic inflammation.
In summary, this chapter highlights the complex composition and inflammatory potential of adipose tissue in obese patients. Hence, it is assumed that the calculation of a person’s BMI by using solely two parameters might lead to misclassifications and excludes important parameters such as percentage body fat (PBF) or rather molecular biomarkers. With regard to the investigations of predispositions for various diseases, it should be considered to include more parameters. In this context, DeLorenzo et al. [30] summarized four obese phenotypes: (1) normal weight obese, (2) metabolically obese normal weight; (3) metabolically healthy obese and (4) metabolically unhealthy obese. Here additional factors concerning body fat composition and distribution (such as fat mass, glucose-levels or CRP-levels) are taken into account. This would also specify the analysis of the impact of obesity on infectious diseases.
3. Adipocytokines Produced by Adipose Tissue
Since the discovery of leptin in 1994, adipose tissue is also known as an endocrine organ, besides its function contributing to energy storage [31]. It can express and secrete a range of proteins, which are termed adipocytokines or adipokines, because of their main but not exclusive production in this specialized type of tissue [18]. In the following section, the main adipokines and their function in inflammatory processes are described. Additionally, we highlight their origin and detected alterations during weight gain (Figure 1).
3.1. Pro-Inflammatory Molecules of the Adipose Tissue
Leptin is the best-characterized adipokine, mainly produced by adipocytes. It was discovered in the 1960s with the help of parabiotic experiments with two mouse strains, the ob/ob (obese) and db/db (diabetes) mutant. This procedure aims at the surgical joining of two mice, leading to the coupling of their blood circulation and enabling the analysis of different physiological or hormonal processes [32]. Both of the investigated mutants result in an obese phenotype. By conjoining individuals of those strains with each other or with lean mice, respectively, different weight changes were observed. Remarkably, the db/db mouse overexpressed a factor but was not able to respond to it with weight loss. In fact, the ob/ob mouse could not produce this factor but reacted to it by losing weight while connected with the db/db mutant or the respective wildtype [33]. 40 years later, this factor and product of the ob gene that seemed to orchestrate the bodyweight of the two mutant strains was named leptin (Greek: leptos = thin) [31]. The gene product of db was termed ObR, the corresponding receptor [34].
An overview of the main downstream branches of leptin are schematically displayed in Figure 2. Leptin is a 16 kDa polypeptide that shows structural similarities to the long-chain cytokines IL-6, IL-12, or G-CSF, all known to contribute to inflammation [35]. The leptin-receptor is a type I cytokine receptor type and exists as six alternatively spliced isoforms with varying intracellular, cytoplasmic parts [36]. One long (ObRb), four short (ObRa, ObRc, ObRd, and ObRf) and one soluble form (ObRe) exist. Only the long isoform possesses the entire intracellular domain, with the conserved tyrosine residues (Y985, Y1077, Y118) [37]. Besides the expression in the hypothalamic region of the brain, where the main function of appetite regulation is orchestrated via leptin signaling, other tissues also express leptin receptor isoforms, including the heart, placenta, liver, muscle, kidney, pancreas, spleen thymus, prostate, testes, ovary, small intestine, and colon [38]. Remarkably, Tsuchiya et al. [39] revealed that the human leptin receptor also occurs in lung tissue.
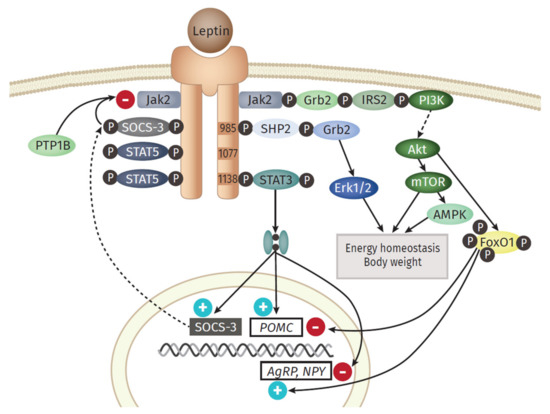
Leptin exerts its function in the hypothalamus via the activation of anorexigenic POMC (Proopiomelanocortin) neurons and orexigenic NPY (Neuropeptide Y)/AgRP (Agouti-related Protein) neurons [40]. The most prominent downstream signaling component of the leptin receptor is the JAK (Janus kinase)/STAT (signal transducer and activator of transcription) pathway [41]. An important downstream gene is SOCS3 (Suppressor of cytokine signaling 3), which itself acts as a potent negative regulator of the leptin signaling, by inhibiting JAK2 [42]. Another signaling branch activated via leptin is the MAPK (Mitogen-activated protein kinase) pathway. Here, the SH2 domain of the phosphatase SH2- containing protein tyrosine phosphatase 2 (SHP2) binds to pY985, becomes phosphorylated by JAKs, which finally activates MAPK extracellular signal-related kinase (ERK1/2) via recruitment of growth factor receptor-bound protein 2 (Grb2) [43]. Leptin also activates Akt by triggering the phosphatidylinositol 3 kinase (PI3K) signaling pathway. Akt inhibits forkhead box O1 (Foxo1), the mediator of the previously mentioned function of leptin in the hypothalamus [44]. Furthermore, downstream of Akt is the Ser/Thr kinase mTOR (mammalian target of rapamycin), a mammalian sensor for the availability of nutrients and stimulator of cell growth, protein biosynthesis, and proliferation [45]. Leptin stimulates the AMPK (adenosine monophosphate-activated protein kinase) pathway, but the outcome differs between the tissues. While activated in hepatocytes and muscle [46], AMPK is inhibited in the hypothalamus, resulting in inhibition of food intake [47].
The observations gained from the parabiosis experiments reflect one main function of leptin: the regulation of food intake and energy expenditure via the leptin-hypothalamus axis. In the hypothalamus, leptin activates anorexigenic neurons, thereby decreasing food intake; at the same time, it leads to the inactivation of orexigenic neurons that stimulate appetite and intake [40]. Counterintuitive to this role in food intake regulation, the level of leptin in the systemic circulation of obese persons is elevated and positively correlated with adipose mass. This hyperleptinemia is a consequence of developing leptin resistance [48]. Several potential explanations for this effect are discussed. On the one hand, the action of a negative feedback loop orchestrated by SOCS3, activated by leptin could serve as a potential link to the leptin resistance. The protein tyrosine phosphatase 1 B (PTP1B) is capable to inhibit the leptin receptor activity by dephosphoryltion of JAK2 [49]. On the other hand, disturbances in the hypothalamic neuronal wiring, impairments in the transport of leptin to the brain, the ObR trafficking, ER stress, or inflammation itself are discussed as potential key events for the development of leptin resistance [36].
Besides its regulatory function in the hypothalamus, leptin itself is defined as a pro-inflammatory adipokine and plays a major role in innate and adaptive immunity [50]. In monocytes, it induces increased production of TNF, IL-6, and ROS as well as cell proliferation [51], thereby stabilizing their activation, phagocytotic activity, and cytokine production [12]. In turn, leptin expression is also elevated by pro-inflammatory cytokines such as TNF and IL-1, indicating a bidirectional interaction between leptin and inflammation [52]. In addition to its interaction with cytokine pathways, leptin stimulates the production of CC-chemokine ligands (CCL3, CCL4, and CCL5) in macrophages [51]. Leptin can also trigger the chemotaxis and ROS production of neutrophils as well as the differentiation, proliferation, activation, and cytotoxicity of natural killer (NK)-cells [53]. Moreover, it inhibits apoptosis and improves the activation and proliferation of T lymphocytes [54]. It also promotes the Th1 phenotype of lymphocytes and the production of IL-2 and IFNγ, while inhibiting the Th2 type and the expression of IL-4 [55].
Another important adipokine is Resistin, belonging to the cysteine-rich family of resistin-like molecules (RELMS) and named according to its connection to the resistance to insulin [56]. The ability to induce insulin resistance is associated with activation of SOCS3, an inhibitor of insulin signaling in adipocytes. However, this has so far only been observed in mice, not in humans [57]. Resistin levels are upregulated in the adipose tissue as well as in the serum of obese individuals [58]. The localization of resistin production seems to differ between mice and men: Whereas the synthesis in humans takes mainly place in macrophages and monocytes [59], it is in mice predominantly produced in adipocytes [60]. Furthermore, the human type only shares a 64% homology with the murine [61]. Nevertheless, it has a known pro-inflammatory effect in humans, since resistin stimulates the expression of TNF and IL-6 in mononuclear cells [62] as well as the expression of pro-inflammatory adhesion molecules vascular cell adhesion protein 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and pentraxin in endothelial cells. Pentraxin directly counteracts adiponectin, an anti-inflammatory adipokine, and fosters the adhesion of leukocytes [63].
Visfatin, also known as nicotinamide phosphoribosyltransferase (NAMPT), represents another type of adipokine, mainly secreted by the visceral type of adipose tissue [64]. In humans, an elevated level of visfatin has been detected in obese and type 2 diabetes patients [65]. Moreover, a positive correlation between IL-6 and the c-reactive protein (CRP) has been observed, linking visfatin to inflammation [64]. In 1987, the first adipokine, adipsin, was described, also known as complement factor D, a part of the alternative activation pathway of the complement system [66]. It is dysregulated in obesity and diabetes models [67]. In obese mice, circulating adipsin levels are decreased [68], whereas in humans a mild elevation could be observed [69]. In 2014, studying mouse models as well as diabetes patients, Lo et al. [70] could demonstrate that adipsin serves a connection between adipocyte function and ß cell physiology in the pancreas.
In this context, a number of studies have demonstrated that interferons (IFNs) are also released from adipose tissue [71]. Controversially, Surendar et al. showing that adiponectin reduces the IFNɣ level but the hypoleptinemia could be shown as responsible for the decrease of the IFNɣ response [72]. IFNɣ influences the function of adipocytes and promotes the inflammation of the adipose tissue [73]. Studies are showing a shift to the Th1-cytokine profile triggered by IFNɣ [74]. Particularly important is in the context of viral pneumonia that the IFN response is the most efficient innate immune response against viral infections [75] The inhibition of the viral replication mediates the antiviral effect, and primarily type I IFN (IFNα/β) plays a crucial role.
Adipocytes and stromovascular cells of the adipose tissue are addtionially able to produce the well-studied pro-inflammatory cytokine TNF [20]. Levels are clearly increased in the systemic circulation and adipose tissue in obese individuals as well as in models of type 2 diabetes [76]. TNF has also been proposed to play an important role in the development of insulin resistance since it debilitates the important tyrosine phosphorylation of the insulin receptor and its substrate IRS1 in muscle and adipose tissue [77].
Nearly one-third of circulating IL-6 is produced in visceral adipose tissue, where it is secreted mainly by macrophages and adipocytes [78]. The plasma levels again positively correlate with obesity in humans [79]. IL-6 also represents a link to insulin resistance, as it has been shown to suppress metabolic processes stimulated by insulin in hepatocytes, possibly induced by SOCS3 expression [80]. Elevated levels of IL-6 are likely connected to an increase in acute phase response proteins, such as CRP [12]. In addition to the above-mentioned cytokines, IL-18 is produced in adipose tissues and shows increased levels in obese individuals [81]. In rodent models, this overexpression leads to a higher amount of cell adhesion molecules, the infiltration of macrophages, and vascular abnormalities [82].
Adipocytes and macrophages are further able to synthesize the retinol-binding protein 4 (RBP4); its levels are elevated under obese conditions and associated with features of the metabolic syndrome in humans [83][84][85]. Lipocalin 2 and angiopoietin-like protein 2 (ANGTPL2) are expressed in adipose tissue and positively correlated with adiposity, hyperglycemia, insulin resistance, and CRP levels in humans [86][87]. Additionally, the chemokines CCL2, also known as monocyte chemoattractant protein-1 (MCP-1) [88], and C-X-C-motif chemokine 5 (CXCL5) [89] are secreted by the adipose tissue. Ob/ob mutant or diet-induced mice (DIO) mice, as well as obese humans, express high levels of MCP-1. In mice, it has already been shown to activate macrophage recruitment and to promote inflammation, glucose intolerance, and insulin insensitivity [90]. Obese and insulin-resistant human individuals also show an increase in CXCL5 levels, a factor that is produced by macrophages within adipose tissue [89].
3.2. Anti-Inflammatory Molecules of the Adipose Tissue
The adipokine with the highest serum levels is adiponectin, which is almost exclusively synthesized by adipocytes [56]. After its discovery in 1995, Hu et al. [91] detected for the first time that obese mice and humans show a downregulated expression of adiponectin in adipose tissue. Conversely to this investigation, especially adipocytes in visceral adipose tissue are the main source of this adipocytokine [92]. It shows structural similarities to the complement factor C1q and is also able to form similar complex structures, such as multimers [93]. Adiponectin has a molecular weight of 30 kDa and accounts for approximately 0.01–0.05% of the plasma protein amount [94]. In the circulation, it is present in different forms, as low-, medium- and high-molecular weight (LMW, MMW, HMW) complexes [95]. Most notably, the HMW type is seen to be the most biologically active isoform [96]. Adiponectin exerts its main functions via the adiponectin receptors 1 and 2 (ADIPOR1 and ADIPOR2), which are expressed in various tissues [97]. The central functions of adiponectin are orchestrated via AMPK signaling [98].
Besides the detected downregulation of adiponectin in subjects with an increased body mass, especially the inverse correlation to glucose intolerance and type 2 diabetes is of importance [99]. In this context, adiponectin appears to promote beta-cell function and survival [100]. Furthermore, it increases insulin sensitivity in hepatocytes [101]. Apart from diabetes, low adiponectin levels are also associated with an increased risk for hepatic fibrosis [102] and cancer [103]. It was also observed, that an elevated level of adiponectin is associated with a decreased susceptibility for myocardial infarction in men [104]. Concerning pulmonary impairments, it is positively associated with lung function in healthy adults [105].
With regard to the inflammatory actions, there are different molecular actions known that suggest adiponectin as a potential antagonist of leptin. Clear anti-inflammatory properties through inhibition of IL-6 production, induction of anti-inflammatory cytokines, such as IL-10 or IL-1 receptor antagonist [106], and reduction in ICAM-1 and VCAM-1 [107] were shown. Secondly, adiponectin levels are decreased in obese and type 2 diabetic subjects and are negatively correlated with visceral mass [108]. Furthermore, the expression seems to be downregulated by pro-inflammatory cytokines TNFα and IL-6 [93], hypoxia, and oxidative stress [109]. It also negatively correlates with the level of CRP in obese or diabetic conditions [93]. Adiponectin can also affect macrophages by stimulating the production of anti-inflammatory cytokines [110]. Along the same line, adiponectin-deficient mice display an increased expression of pro-inflammatory M1 type markers and decreased anti-inflammatory M2 type markers [111].
In 2010, another anti-inflammatory adipokine, secreted frizzled-related protein 5 (SFRP5), was discovered [112]. The level of SFRP5 is downregulated in adipose tissue from obese rodents as well as from obese humans with insulin resistance [50]. A deficiency of this adipokine leads to an accumulation of macrophages resulting in increased pro-inflammatory cytokine production [50]. A clinical study furthermore demonstrated an association between lower SFRP5 levels in adults with impaired glucose intolerance and type 2 diabetes, as well as a negative correlation to increased BMI [113].
The overall dysregulation of secreted adipocytokines creates a low-grade chronically inflamed environment during weight gain conditions. This chronic inflammatory state is closely linked to the predisposition to various comorbidities of obesity: lower levels of adiponectin, for example, are known to lead to an elevated risk to develop cardiovascular diseases, such as hypertension [114] or myocardial infarction [104]. This dysregulation is further connected to the development of insulin resistance [115] and potentially impacts the comorbidities of cancer and asthma [18]. Moreover, a connection between this chronic state of low-grade inflammation and the susceptibility to viral or bacterial pulmonary infections seems likely. In Chapter 5, we summarize, connect, and compare different publications on pulmonary infections, the most frequent infection focus, and discuss obesity as a risk factor for severe infections. Different experimental setups to mimic obesity in vivo and in vitro are summarized in Chapter 4 beforehand.
References
- Gray, D.S. Diagnosis and Prevalence of Obesity. Med. Clin. N. Am. 1989, 73, 1–13.
- Rosalki, S.B. The Clinical Biochemistry of Alcohol. In Scientific Foundations of Biochemistry in Clinical Practice; Elsevier: Amsterdam, The Netherlands, 1994; pp. 121–143.
- Obesity and Overweight. Available online: (accessed on 7 November 2020).
- Quetelet, L.A. A Treatise on Man and the Development of His Faculties. 1842. Obes. Res. 1994, 2, 72–85.
- Gadde, K.M.; Martin, C.K.; Berthoud, H.R.; Heymsfield, S.B. Obesity: Pathophysiology and Management. J. Am. Coll. Cardiol. 2018, 71, 69–84.
- Haslam, D.W.; James, W.P.T. Obesity. Lancet 2005, 366, 1197–1209.
- Koenig, S.M. Pulmonary Complications of Obesity. Am. J. Med. Sci. 2001, 321, 249–279.
- Robinson, J.; Swift-Scanlan, T.; Salyer, J.; Jones, T. The Obesity Paradox in Sepsis: A Theoretical Framework. Biol. Res. Nurs. 2020, 22, 287–294.
- Jackson-Morris, A.M.; Nugent, R.; Ralston, J.; Barata Cavalcante, O.; Wilding, J. Strengthening Resistance to the COVID-19 Pandemic and Fostering Future Resilience Requires Concerted Action on Obesity. Glob. Health Action 2020, 13.
- Noor, F.M.; Islam, M.M. Prevalence and Associated Risk Factors of Mortality Among COVID-19 Patients: A Meta-Analysis. J. Community Health 2020, 45, 1270–1282.
- Lavie, C.J.; Milani, R.; Ventura, H.O. Obesity, Heart Disease, and Favorable Prognosis-Truth or Paradox? Am. J. Med. 2007.
- Fantuzzi, G. Adipose Tissue, Adipokines, and Inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919.
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected Evidence for Active Brown Adipose Tissue in Adult Humans. Am. J. Physiol. Endocrinol. Metab. 2007.
- Cinti, S. White, Brown, Beige and Pink: A Rainbow in the Adipose Organ. Curr. Opin. Endocr. Metab. Res. 2019, 4, 29–36.
- Himms-Hagen, J.; Melnyk, A.; Zingaretti, M.C.; Ceresi, E.; Barbatelli, G.; Cinti, S. Multilocular Fat Cells in WAT of CL-316243-Treated Rats Derive Directly from White Adipocytes. Am. J. Physiol. Cell Physiol. 2000, 279.
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-Dependent Myokine That Drives Brown-Fat-like Development of White Fat and Thermogenesis. Nature 2012, 481, 463–468.
- Giordano, A.; Smorlesi, A.; Frontini, A.; Barbatelli, G.; Cint, S. White, Brown and Pink Adipocytes: The Extraordinary Plasticity of the Adipose Organ. Eur. J. Endocrinol. 2014.
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators Linking Adipose Tissue, Inflammation and Immunity. Nat. Rev. Immunol. 2006, 6, 772–783.
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Invest. 2003, 112, 1796–1808.
- Frayn, K.N.; Karpe, F.; Fielding, B.A.; Macdonald, I.A.; Coppack, S.W. Integrative Physiology of Human Adipose Tissue. Int. J. Obes. 2003, 27, 875–888.
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, Stress, and Diabetes. J. Clin. Invest. 2005, 115, 1111–1119.
- Calle, E.E.; Kaaks, R. Overweight, Obesity and Cancer: Epidemiological Evidence and Proposed Mechanisms. Nat. Rev. Cancer 2004, 579–591.
- Goossens, G.H. The Role of Adipose Tissue Dysfunction in the Pathogenesis of Obesity-Related Insulin Resistance. Physiol. Behav. 2008, 94, 206–218.
- Rosen, E.D.; Spiegelman, B.M. What We Talk about When We Talk about Fat. Cell 2014, 20–44.
- Virtue, S.; Vidal-Puig, A. Adipose Tissue Expandability, Lipotoxicity and the Metabolic Syndrome-An Allostatic Perspective. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 338–349.
- Sun, K.; Scherer, P.E. Adipose Tissue Dysfunction: A Multistep Process; Springer: Berlin/Heidelberg, Germany, 2010; pp. 67–75.
- Sun, K.; Kusminski, C.M.; Scherer, P.E.; Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose Tissue Remodeling and Obesity. J. Clin. Investig. 2011, 121, 2094–2101.
- Wellen, K.E.; Hotamisligil, G.S. Obesity-Induced Inflammatory Changes in Adipose Tissue. J. Clin. Investig. 2003, 1785–1788.
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R.; Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization Find the Latest Version: Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization. J Clin Invest 2007, 117, 175–184.
- De Lorenzo, A.; Soldati, L.; Sarlo, F.; Calvani, M.; Di Lorenzo, N.; Di Renzo, L. New Obesity Classification Criteria as a Tool for Bariatric Surgery Indication. World J. Gastroenterol. 2016, 22, 681–703.
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional Cloning of the Mouse Obese Gene and Its Human Homologue. Nature 1994, 372, 425–432.
- Kamran, P.; Sereti, K.I.; Zhao, P.; Ali, S.R.; Weissman, I.L.; Ardehali, R. Parabiosis in Mice: A Detailed Protocol. J. Vis. Exp. 2013, e50556.
- Coleman, D.L. A Historical Perspective on Leptin. Nat. Med. 2010, 1097–1099.
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and Expression Cloning of a Leptin Receptor, OB-R. Cell 1995, 83, 1263–1271.
- La Cava, A.; Matarese, G. The Weight of Leptin in Immunity. Nat. Rev. Immunol. 2004, 4, 371–379.
- Wauman, J.; Zabeau, L.; Tavernier, J. The Leptin Receptor Complex: Heavier than Expected? Front. Endocrinol. 2017, 8, 1–20.
- Haniu, M.; Arakawa, T.; Bures, E.J.; Young, Y.; Hui, J.O.; Rohde, M.F.; Welcher, A.A.; Horan, T. Human Leptin Receptor: Determination of Disulfide Structure and N- Glycosylation Sites of the Extracellular Domain. J. Biol. Chem. 1998, 273, 28691–28699.
- Kielar, D.; Clark, J.S.C.; Ciechanowicz, A.; Kurzawski, G.; Sulikowski, T.; Naruszewicz, M. Leptin Receptor Isoforms Expressed in Human Adipose Tissue. Metabolism 1998, 47, 844–847.
- Tsuchiya, T.; Shimizu, H.; Horie, T.; Mori, M. Expression of Leptin Receptor in Lung: Leptin as a Growth Factor. Eur. J. Pharmacol. 1999, 365, 273–279.
- Hahn, T.M.; Breininger, J.F.; Baskin, D.G.; Schwartz, M.W. Coexpression of Agrp and NPY in Fasting-Activated Hypothalamic Neurons. Nat. Neurosci. 1998, 1, 271–272.
- Vaisse, C.; Halaas, J.L.; Horvath, C.M.; Darnell, J.E.; Stoffel, M.; Friedman, J.M. Leptin Activation of Stat3 in the Hypothalamus of Wild–Type and Ob/Ob Mice but Not Db/Db Mice. Nat. Genet. 1996, 14, 95–97.
- Bjørbaek, C.; Elmquist, J.K.; Daniel Frantz, J.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a Potential Mediator of Central Leptin Resistance Cytokine-like Signal Transduction by Stimulating the JAK-STAT Pathway via the Long Leptin Receptor Isoform. Mol. Cell 1998, 1, 619–625.
- Bjørbæk, C.; Buchholz, R.M.; Davis, S.M.; Bates, S.H.; Pierroz, D.D.; Gu, H.; Neel, B.G.; Myers, M.G.; Flier, J.S. Divergent Roles of SHP-2 in ERK Activation by Leptin Receptors. J. Biol. Chem. 2001, 276, 4747–4755.
- Kitamura, T. Forkhead Protein FoxO1 Mediates Agrp-Dependent Effects of Leptin on Food Intake. Nat Med. 2006, 12, 534–540.
- Cota, D.; Proulx, K.; Blake Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic MTOR Signaling Regulates Food Intake. Science 2006, 312, 927–930.
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin Stimulates Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nature 2002, 415, 339–343.
- Minokoshi, Y.; Alquier, T.; Furukawa, H.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-Kinase Regulates Food Intake by Responding to Hormonal and Nutrient Signals in the Hypothalamus. Nature 2004, 428, 569–574.
- Wauman, J.; Tavernier, J. Leptin Receptor Signaling: Pathways to Leptin Resistance. Front. Biosci. 2011, 16, 2771–2793.
- Kaszubska, W.; Falls, H.D.; Schaefer, V.G.; Haasch, D.; Frost, L.; Hessler, P.; Kroeger, P.E.; White, D.W.; Jirousek, M.R.; Trevillyan, J.M. Protein Tyrosine Phosphatase 1B Negatively Regulates Leptin Signaling in a Hypothalamic Cell Line. Mol. Cell. Endocrinol. 2002, 195, 109–118.
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in Inflammation and Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 85.
- Santos-Alvarez, J.; Goberna, R.; Sá Nchez-Margalet, V. Human Leptin Stimulates Proliferation and Activation of Human Circulating Monocytes. Cell Immunol. 1999, 6–11.
- Paz-Filho, G.; Mastronardi, C.; Franco, C.B.; Wang, K.B.; Wong, M.-L.; Licinio, J. Leptin: Molecular Mechanisms, Systemic pro-Inflammatory Effects, and Clinical Implications. Arq. Bras. Endocrinol. Metabol. 2012, 56, 597–607.
- Tian, Z.; Sun, R.; Wei, H.; Gao, B. Impaired Natural Killer (NK) Cell Activity in Leptin Receptor Deficient Mice: Leptin as a Critical Regulator in NK Cell Development and Activation. Biochem. Biophys. Res. Commun. 2002, 298, 297–302.
- La Cava, A.; Alviggi, C.; Matarese, G. Unraveling the Multiple Roles of Leptin in Inflammation and Autoimmunity. J. Mol. Med. 2004, 4–11.
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin Modulates the T-Cell Immune Response and Reverses Starvation-Induced Immunosuppression. Nature 1998, 394, 897–901.
- Bełtowski, J. Adiponectin and Resistin-New Hormones of White Adipose Tissue. Med Sci. Monitor. 2003, 9, RA55–RA61.
- Steppan, C.M.; Wang, J.; Whiteman, E.L.; Birnbaum, M.J.; Lazar, M.A. Activation of SOCS-3 by Resistin. Mol. Cell. Biol. 2005, 25, 1569–1575.
- Degawa-Yamauchi, M.; Bovenkerk, J.E.; Juliar, B.E.; Watson, W.; Kerr, K.; Jones, R.; Zhu, Q.; Considine, R.V. Serum Resistin (FIZZ3) Protein Is Increased in Obese Humans. J. Clin. Endocrinol. Metab. 2003, 88, 5452–5455.
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The Hormone Resistin Links Obesity to Diabetes. Nature 2001, 409, 307–312.
- Savage, D.B.; Sewter, C.P.; Klenk, E.S.; Segal, D.G.; Vidal-Puig, A.; Considine, R.V.; O’Rahilly, S. Resistin / Fizz3 Expression in Relation to Obesity and Peroxisome Proliferator-Activated Receptor-γ Action in Humans. Diabetes 2001, 50, 2199–2202.
- Banerjee, R.R.; Lazar, M.A. Resistin: Molecular History and Prognosis. J. Mol. Med. 2003, 218–226.
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an Adipokine with Potent Proinflammatory Properties. J. Immunol. 2005, 174, 5789–5795.
- Verma, S.; Li, S.H.; Wang, C.H.; Fedak, P.W.M.; Li, R.K.; Weisel, R.D.; Mickle, D.A.G. Resistin Promotes Endothelial Cell Activation: Further Evidence of Adipokine-Endothelial Interaction. Circulation 2003, 108, 736–740.
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin Regulates Insulin Secretion in β Cells as a Systemic NAD Biosynthetic Enzyme. Cell Metab. 2007, 6, 363–375.
- Haider, D.G.; Schindler, K.; Schaller, G.; Prager, G.; Wolzt, M.; Ludvik, B. Increased Plasma Visfatin Concentrations in Morbidly Obese Subjects Are Reduced after Gastric Banding. J. Clin. Endocrinol. Metab. 2006, 91, 1578–1581.
- Cook, K.S.; Min, H.Y.; Johnson, D.; Chaplinsky, R.J.; Flier, J.S.; Hunt, C.R.; Spiegelman, B.M. Adipsin: A Circulating Serine Protease Homolog Secreted by Adipose Tissue and Sciatic Nerve. Science 1987, 237, 402–405.
- White, R.T.; Damm, D.; Hancock, N.; Rosen, B.S.; Lowell, B.B.; Usher, P.; Flier, J.S.; Spiegelman, B.M. Human Adipsin Is Identical to Complement Factor D and Is Expressed at High Levels in Adipose Tissue. J. Biol. Chem. 1992, 267, 9210–9213.
- Flier, J.S.; Cook, K.S.; Usher, P.; Spiegelman, B.M. Severely Impaired Adipsin Expression in Genetic and Acquired Obesity. Science 1987, 237, 405–408.
- Napolitano, A.; Lowell, B.B.; Damm, D.; Leibel, R.L.; Ravussin, E.; Jimerson, D.C.; Lesem, M.D.; Van Dyke, D.C.; Daly, P.A.; Chatis, P.; et al. Concentrations of Adipsin in Blood and Rates of Adipsin Secretion by Adipose Tissue in Humans with Normal, Elevated and Diminished Adipose Tissue Mass. Int. J. Obes. 1994, 18, 213–218.
- Lo, J.C.; Ljubicic, S.; Leibiger, B.; Kern, M.; Leibiger, I.B.; Moede, T.; Kelly, M.E.; Chatterjee Bhowmick, D.; Murano, I.; Cohen, P.; et al. Adipsin is an Adipokine That Improves β Cell Function in Diabetes. Cell 2014, 158, 41–53.
- Wentworth, J.M.; Zhang, J.G.; Bandala-Sanchez, E.; Naselli, G.; Liu, R.; Ritchie, M.; Smyth, G.K.; O’Brien, P.E.; Harrison, L.C. Interferon-Gamma Released from Omental Adipose Tissue of Insulin-Resistant Humans Alters Adipocyte Phenotype and Impairs Response to Insulin and Adiponectin Release. Int. J. Obes. 2017, 41, 1782–1789.
- Surendar, J.; Frohberger, S.J.; Karunakaran, I.; Schmitt, V.; Stamminger, W.; Neumann, A.-L.; Wilhelm, C.; Hoerauf, A.; Hübner, M.P. Adiponectin Limits IFN-γ and IL-17 Producing CD4 T Cells in Obesity by Restraining Cell Intrinsic Glycolysis. Front. Immunol. 2019, 10, 2555.
- Rocha, V.Z.; Libby, P. Obesity, Inflammation, and Atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409.
- Pacifico, L.; Di Renzo, L.; Anania, C.; Osborn, J.F.; Ippoliti, F.; Schiavo, E.; Chiesa, C. Increased T-Helper Interferon-γ-Secreting Cells in Obese Children. Eur. J. Endocrinol. 2006, 154, 691–697.
- Thiel, V.; Weber, F. Interferon and Cytokine Responses to SARS-Coronavirus Infection. Cytokine Growth Factor Rev. 2008, 121–132.
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The Expression of Tumor Necrosis Factor in Human Adipose Tissue: Regulation by Obesity, Weight Loss, and Relationship to Lipoprotein Lipase. J. Clin. Invest. 1995, 95, 2111–2119.
- Hotamisligil, G.S.; Budavari, A.; Murray, D.; Spiegelman, B.M. Reduced Tyrosine Kinase Activity of the Insulin Receptor in Obesity- Diabetes. Central Role of Tumor Necrosis Factor-α. J. Clin. Invest. 1994, 94, 1543–1549.
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the Release of Adipokines by Adipose Tissue, Adipose Tissue Matrix, and Adipocytes from Visceral and Subcutaneous Abdominal Adipose Tissues of Obese Humans. Endocrinology 2004, 145, 2273–2282.
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and Subcutaneous Adipose Tissues of Obese Subjects Release Interleukin-6: Depot Difference and Regulation by Glucocorticoid 1. J. Clin. Endocrinol. Metab. 1998, 83, 847–850.
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of Cytokine Signaling-3 (SOCS-3), a Potential Mediator of Interleukin-6-Dependent Insulin Resistance in Hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746.
- Esposito, K.; Pontillo, A.; Ciotola, M.; Di Palo, C.; Grella, E.; Nicoletti, G.; Giugliano, D. Weight Loss Reduces Interleukin-18 Levels in Obese Women. J. Clin. Endocrinol. Metab. 2002, 87, 3864–3866.
- Tan, H.W.; Liu, X.; Bi, X.P.; Xing, S.S.; Li, L.; Gong, H.P.; Zhong, M.; Wang, Z.H.; Zhang, Y.; Zhang, W. IL-18 Overexpression Promotes Vascular Inflammation and Remodeling in a Rat Model of Metabolic Syndrome. Atherosclerosis 2010, 208, 350–357.
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum Retinol Binding Protein 4 Contributes to Insulin Resistance in Obesity and Type 2 Diabetes. Nature 2005, 436, 356–362.
- Graham, T.E.; Yang, Q.; Blüher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.-A.; Smith, U.; et al. Retinol-Binding Protein 4 and Insulin Resistance in Lean, Obese, and Diabetic Subjects. N. Engl. J. Med. 2006, 354, 2552–2563.
- Broch, M.; Ramírez, R.; Auguet, M.T.; Alcaide, M.J.; Aguilar, C.; Garcia-España, A.; Richart, C.; Xxiii, C.; Guasch, M. Macrophages Are Novel Sites of Expression and Regulation of Retinol Binding Protein-4 (RBP4). Physiol. Res. 2010, 59, 299–303.
- Tabata, M.; Kadomatsu, T.; Fukuhara, S.; Miyata, K.; Ito, Y.; Endo, M.; Urano, T.; Zhu, H.J.; Tsukano, H.; Tazume, H.; et al. Angiopoietin-like Protein 2 Promotes Chronic Adipose Tissue Inflammation and Obesity-Related Systemic Insulin Resistance. Cell Metab. 2009, 10, 178–188.
- Wang, Y.; Lam, K.S.L.; Kraegen, E.W.; Sweeney, G.; Zhang, J.; Tso, A.W.; Chow, W.-S.; Wat, N.M.; Xu, J.Y.; Hoo, R.L.; et al. Lipocalin-2 Is an Inflammatory Marker Closely Associated with Obesity, Insulin Resistance, and Hyperglycemia in Humans. Clin. Chem. 2007, 53, 34–41.
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 Contributes to Macrophage Infiltration into Adipose Tissue, Insulin Resistance, and Hepatic Steatosis in Obesity. J. Clin. Invest. 2006, 116, 1494–1505.
- Chavey, C.; Lazennec, G.; Lagarrigue, S.; Clapé, C.; Iankova, I.; Teyssier, J.; Annicotte, J.S.; Schmidt, J.; Mataki, C.; Yamamoto, H.; et al. CXC Ligand 5 Is an Adipose-Tissue Derived Factor That Links Obesity to Insulin Resistance. Cell Metab. 2009, 9, 339–349.
- Sartipy, P.; Loskutoff, D.J. Monocyte Chemoattractant Protein 1 in Obesity and Insulin Resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270.
- Hu, E.; Liang, P.; Spiegelman, B.M. AdipoQ Is a Novel Adipose-Specific Gene Dysregulated in Obesity. J. Biol. Chem. 1996, 271, 10697–10703.
- Steffes, M.W.; Gross, M.D.; Schreiner, P.J.; Yu, X.; Hilner, J.E.; Gingerich, R.; Jacobs, D.R. Serum Adiponectin in Young Adults-Interactions with Central Adiposity, Circulating Levels of Glucose, and Insulin Resistance: The CARDIA Study. Ann. Epidemiol. 2004, 14, 492–498.
- Ouchi, N.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Walsh, K. Obesity, Adiponectin and Vascular Inflammatory Disease. Curr. Opin. Lipidol. 2003, 14, 561–566.
- Pischon, T.; Hotamisligil, G.S.; Rimm, E.B. Adiponectin: Stability in Plasma over 36 Hours and within-Person Variation over 1 Year. Clin. Chem. 2003, 49, 650–652.
- Waki, H.; Yamauchi, T.; Kamon, J.; Ito, Y.; Uchida, S.; Kita, S.; Hara, K.; Hada, Y.; Vasseur, F.; Froguel, P.; et al. Impaired Multimerization of Human Adiponectin Mutants Associated with Diabetes. Molecular Structure and Multimer Formation of Adiponectin. J. Biol. Chem. 2003, 278, 40352–40363.
- Hara, K.; Horikoshi, M.; Yamauchi, T.; Yago, H.; Miyazaki, O.; Ebinuma, H.; Imai, Y.; Nagai, R.; Kadowaki, T. Measurement of the High-Molecular Weight Form of Adiponectin in Plasma Is Useful for the Prediction of Insulin Resistance and Metabolic Syndrome. Diabetes Care 2006, 29, 1357–1362.
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of Adiponectin Receptors That Mediate Antidiabetic Metabolic Effects. Nature 2003, 423, 762–769.
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin Stimulates Glucose Utilization and Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nat. Med. 2002, 8, 1288–1295.
- Turer, A.T.; Khera, A.; Ayers, C.R.; Turer, C.B.; Grundy, S.M.; Vega, G.L.; Scherer, P.E. Adipose Tissue Mass and Location Affect Circulating Adiponectin Levels. Diabetologia 2011, 54, 2515–2524.
- Wijesekara, N.; Krishnamurthy, M.; Bhattacharjee, A.; Suhail, A.; Sweeney, G.; Wheeler, M.B. Adiponectin-Induced ERK and Akt Phosphorylation Protects against Pancreatic Beta Cell Apoptosis and Increases Insulin Gene Expression and Secretion. J. Biol. Chem. 2010, 285, 33623–33631.
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The Adipocyte-Secreted Protein Acrp30 Enhances Hepatic Insulin Action. Nat. Med. 2001, 7, 947–953.
- Schäffler, A.; Schölmerich, J.; Büchler, C. Mechanisms of Disease: Adipocytokines and Visceral Adipose Tissue-Emerging Role in Nonalcoholic Fatty Liver Disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 273–280.
- Kelesidis, I.; Kelesidis, T.; Mantzoros, C.S. Adiponectin and Cancer: A Systematic Review. Br. J. Cancer 2006, 1221–1225.
- Pischon, T.; Girman, C.J.; Hotamisligil, G.S.; Rifai, N.; Hu, F.B.; Rimm, E.B. Plasma Adiponectin Levels and Risk of Myocardial Infarction in Men. J. Am. Med. Assoc. 2004, 291, 1730–1737.
- Thyagarajan, B.; Jacobs, D.R.; Smith, L.J.; Kalhan, R.; Gross, M.D.; Sood, A. Serum Adiponectin Is Positively Associated with Lung Function in Young Adults, Independent of Obesity: The CARDIA Study. Respir. Res. 2010, 11.
- Wulster-Radcliffe, M.C.; Ajuwon, K.M.; Wang, J.; Christian, J.A.; Spurlock, M.E. Adiponectin Differentially Regulates Cytokines in Porcine Macrophages. Biochem. Biophys. Res. Commun. 2004, 316, 924–929.
- Ouchi, N.; Kihara, S.; Arita, Y.; Maeda, K.; Kuriyama, H.; Okamoto, Y.; Hotta, K.; Nishida, M.; Takahashi, M.; Nakamura, T.; et al. Novel Modulator for Endothelial Adhesion Molecules: Adipocyte-Derived Plasma Protein Adiponectin. Circulation 1999, 100, 2473–2476.
- Ryo, M.; Nakamura, T.; Kihara, S.; Kumada, M.; Shibazaki, S.; Takahashi, M.; Nagai, M.; Matsuzawa, Y.; Funahashi, T. Adiponectin as a Biomarker of the Metabolic Syndrome. Circ. J. 2004, 68, 975–981.
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose Tissue Hypoxia in Obesity and Its Impact on Adipocytokine Dysregulation. Diabetes 2007, 56, 901–911.
- Kumada, M.; Kihara, S.; Ouchi, N.; Kobayashi, H.; Okamoto, Y.; Ohashi, K.; Maeda, K.; Nagaretani, H.; Kishida, K.; Maeda, N.; et al. Adiponectin Specifically Increased Tissue Inhibitor of Metalloproteinase-1 Through Interleukin-10 Expression in Human Macrophages. Circulation 2004, 109, 2046–2049.
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin Promotes Macrophage Polarization toward an Anti-Inflammatory Phenotype. J. Biol. Chem. 2010, 285, 6153–6160.
- Ouchi, N.; Higuchi, A.; Ohashi, K.; Oshima, Y.; Gokce, N.; Shibata, R.; Akasaki, Y.; Shimono, A.; Walsh, K. Sfrp5 Is an Anti-Inflammatory Adipokine That Modulates Metabolic Dysfunction in Obesity. Science 2010, 329, 454–457.
- Hu, Z.; Deng, H.; Qu, H. Plasma SFRP5 Levels Are Decreased in Chinese Subjects with Obesity and Type 2 Diabetes and Negatively Correlated with Parameters of Insulin Resistance. Diabetes Res. Clin. Pract. 2013, 99, 391–395.
- Iwashima, Y.; Katsuya, T.; Ishikawa, K.; Ouchi, N.; Ohishi, M.; Sugimoto, K.; Fu, Y.; Motone, M.; Yamamoto, K.; Matsuo, A.; et al. Hypoadiponectinemia Is an Independent Risk Factor for Hypertension. Hypertension 2004, 43, 1318–1323.
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose Expression of Tumor Necrosis Factor-α: Direct Role in Obesity-Linked Insulin Resistance. Science 1993, 259, 87–91.


