 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Valentinovna Bulankina | + 4803 word(s) | 4803 | 2021-05-14 05:23:11 | | | |
| 2 | Vivi Li | Meta information modification | 4803 | 2021-05-17 05:02:40 | | |
Video Upload Options
Ferlins are multiple-C2-domain proteins involved in Ca2+-triggered membrane dynamics within the secretory, endocytic and lysosomal pathways. In bony vertebrates there are six ferlin genes encoding, in humans, dysferlin, otoferlin, myoferlin, Fer1L5 and 6 and the long noncoding RNA Fer1L4. Mutations in DYSF (dysferlin) can cause a range of muscle diseases with various clinical manifestations collectively known as dysferlinopathies, including limb-girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi myopathy. A mutation in MYOF (myoferlin) was linked to a muscular dystrophy accompanied by cardiomyopathy. Mutations in OTOF (otoferlin) can be the cause of nonsyndromic deafness DFNB9. Dysregulated expression of any human ferlin may be associated with development of cancer.
1. Introduction
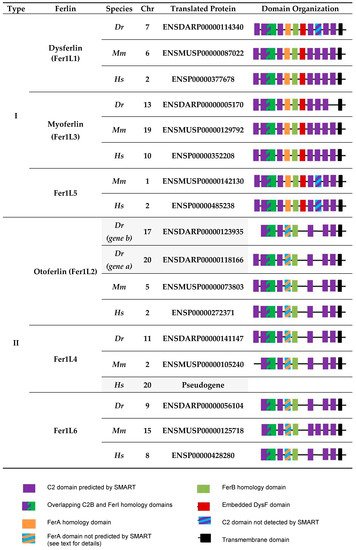
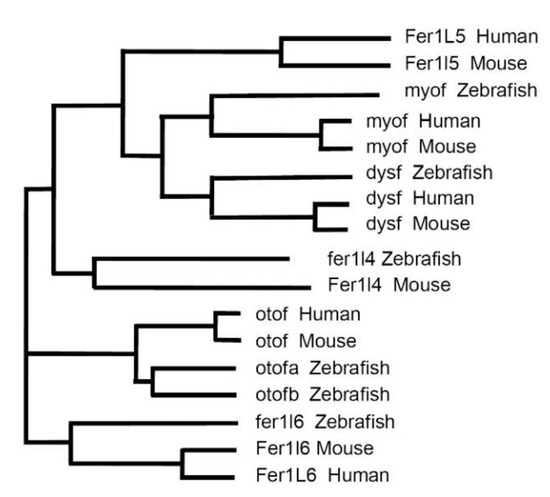
2. Vertebrate Ferlins: Family Members and Domain Organization
3. Functions of Dysferlin in Muscle
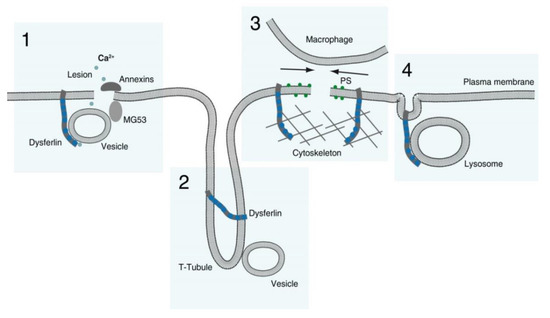
3.1. Dysferlin Functions in Sarcolemma Repair
-
Membranous repair patch or plug formation;
-
T-tubule stabilization (see below) with T-tubule as a possible membrane reservoir;
-
PS-sorting; recruitment of macrophages and contraction of the membrane wound, and;
-
Lysosome exocytosis.
3.2. Dysferlin Functions in Triad Biology
3.3. Dysferlin in the Differentiation, Growth and Regeneration of Skeletal Muscle
4. Functions of Myoferlin and Fer1L5
5. Ferlins in Human Diseases: Dysferlinopathies and Their Pathomechanisms
5.1. Defective Repair of Myofiber Sarcolemma and, Possibly, T-Tubules
5.2. Changes in Muscle Fibers Ca2+ Homeostasis
5.3. Impaired Muscle Growth and Regeneration
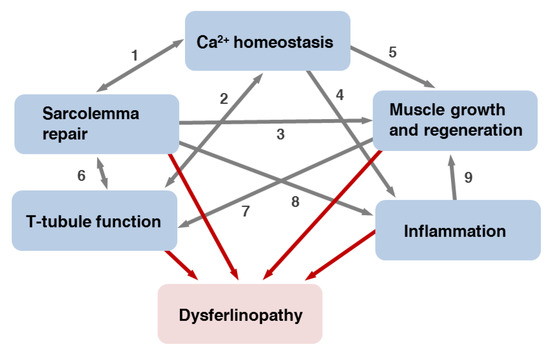
5.4. Inflammatory Processes
References
- Hofhuis, J.; Bersch, K.; Büssenschütt, R.; Drzymalski, M.; Liebetanz, D.; Nikolaev, V.O.; Wagner, S.; Maier, L.S.; Gärtner, J.; Klinge, L.; et al. Dysferlin mediates membrane tubulation and links T-tubule biogenesis to muscular dystrophy. J. Cell. Sci. 2017, 130, 841–852.
- Johnson, C.P. Emerging Functional Differences between the Synaptotagmin and Ferlin Calcium Sensor Families. Biochemistry 2017, 56, 6413–6417.
- Pangrsic, T.; Vogl, C. Balancing presynaptic release and endocytic membrane retrieval at hair cell ribbon synapses. FEBS Lett. 2018, 592, 3633–3650.
- Bashir, R.; Britton, S.; Strachan, T.; Keers, S.; Vafiadaki, E.; Lako, M.; Richard, I.; Marchand, S.; Bourg, N.; Argov, Z.; et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998, 20, 37–42.
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36.
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat. Genet. 1999, 21, 363–369.
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Sergushichev, A.; Dmitrieva, R.; Khudiakov, A.; Jorholt, J.; Smolina, N.; Sukhareva, K.; Fomicheva, Y.; et al. Truncating Variant in Myof Gene Is Associated With Limb-Girdle Type Muscular Dystrophy and Cardiomyopathy. Front. Genet. 2019, 10, 608.
- Peulen, O.; Rademaker, G.; Anania, S.; Turtoi, A.; Bellahcène, A.; Castronovo, V. Ferlin Overview: From Membrane to Cancer Biology. Cells 2019, 8, E954.
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751.
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432.
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496.
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641.
- Lek, A.; Lek, M.; North, K.N.; Cooper, S.T. Phylogenetic analysis of ferlin genes reveals ancient eukaryotic origins. BMC Evol. Biol. 2010, 10, 231.
- Song, H.; Sun, W.; Ye, G.; Ding, X.; Liu, Z.; Zhang, S.; Xia, T.; Xiao, B.; Xi, Y.; Guo, J. Long non-coding RNA expression profile in human gastric cancer and its clinical significances. J. Transl. Med. 2013, 11, 225.
- Redpath, G.M.I.; Sophocleous, R.A.; Turnbull, L.; Whitchurch, C.B.; Cooper, S.T. Ferlins Show Tissue-Specific Expression and Segregate as Plasma Membrane/Late Endosomal or Trans-Golgi/Recycling Ferlins. Traffic 2016, 17, 245–266.
- Chatterjee, P.; Padmanarayana, M.; Abdullah, N.; Holman, C.L.; LaDu, J.; Tanguay, R.L.; Johnson, C.P. Otoferlin deficiency in zebrafish results in defects in balance and hearing: Rescue of the balance and hearing phenotype with full-length and truncated forms of mouse otoferlin. Mol. Cell. Biol. 2015, 35, 1043–1054.
- Bonventre, J.A.; Holman, C.; Manchanda, A.; Codding, S.J.; Chau, T.; Huegel, J.; Barton, C.; Tanguay, R.; Johnson, C.P. Fer1l6 is essential for the development of vertebrate muscle tissue in zebrafish. Mol. Biol. Cell 2019, 30, 293–301.
- Harsini, F.M.; Chebrolu, S.; Fuson, K.L.; White, M.A.; Rice, A.M.; Sutton, R.B. FerA is a Membrane-Associating Four-Helix Bundle Domain in the Ferlin Family of Membrane-Fusion Proteins. Sci. Rep. 2018, 8, 10949.
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172.
- Barthélémy, F.; Defour, A.; Lévy, N.; Krahn, M.; Bartoli, M. Muscle Cells Fix Breaches by Orchestrating a Membrane Repair Ballet. J. Neuromuscul. Dis. 2018, 5, 21–28.
- Abdullah, N.; Padmanarayana, M.; Marty, N.J.; Johnson, C.P. Quantitation of the calcium and membrane binding properties of the C2 domains of dysferlin. Biophys. J. 2014, 106, 382–389.
- Therrien, C.; Di Fulvio, S.; Pickles, S.; Sinnreich, M. Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry 2009, 48, 2377–2384.
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473.
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718.
- Hernández-Deviez, D.J.; Howes, M.T.; Laval, S.H.; Bushby, K.; Hancock, J.F.; Parton, R.G. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J. Biol. Chem. 2008, 283, 6476–6488.
- Cai, C.; Weisleder, N.; Ko, J.-K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902.
- Huang, Y.; Laval, S.H.; van Remoortere, A.; Baudier, J.; Benaud, C.; Anderson, L.V.B.; Straub, V.; Deelder, A.; Frants, R.R.; den Dunnen, J.T.; et al. AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J. 2007, 21, 732–742.
- Park, J.W.; Kim, I.Y.; Choi, J.W.; Lim, H.J.; Shin, J.H.; Kim, Y.N.; Lee, S.H.; Son, Y.; Sohn, M.; Woo, J.K.; et al. AHNAK Loss in Mice Promotes Type II Pneumocyte Hyperplasia and Lung Tumor Development. Mol. Cancer Res. 2018, 16, 1287–1298.
- de Morrée, A.; Hensbergen, P.J.; van Haagen, H.H.H.B.M.; Dragan, I.; Deelder, A.M.; ’t Hoen, P.A.C.; Frants, R.R.; van der Maarel, S.M. Proteomic analysis of the dysferlin protein complex unveils its importance for sarcolemmal maintenance and integrity. PLoS ONE 2010, 5, e13854.
- Matsuda, C.; Kameyama, K.; Tagawa, K.; Ogawa, M.; Suzuki, A.; Yamaji, S.; Okamoto, H.; Nishino, I.; Hayashi, Y.K. Dysferlin interacts with affixin (beta-parvin) at the sarcolemma. J. Neuropathol. Exp. Neurol. 2005, 64, 334–340.
- Flix, B.; de la Torre, C.; Castillo, J.; Casal, C.; Illa, I.; Gallardo, E. Dysferlin interacts with calsequestrin-1, myomesin-2 and dynein in human skeletal muscle. Int. J. Biochem. Cell Biol. 2013, 45, 1927–1938.
- Defour, A.; Van der Meulen, J.H.; Bhat, R.; Bigot, A.; Bashir, R.; Nagaraju, K.; Jaiswal, J.K. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014, 5, e1306.
- McDade, J.R.; Archambeau, A.; Michele, D.E. Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair. FASEB J. 2014, 28, 3660–3670.
- Middel, V.; Zhou, L.; Takamiya, M.; Beil, T.; Shahid, M.; Roostalu, U.; Grabher, C.; Rastegar, S.; Reischl, M.; Nienhaus, G.U.; et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat Commun 2016, 7, 12875.
- Moe, A.M.; Golding, A.E.; Bement, W.M. Cell healing: Calcium, repair and regeneration. Semin. Cell Dev. Biol. 2015, 45, 18–23.
- Roostalu, U.; Strähle, U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev. Cell 2012, 22, 515–529.
- Piccolo, F.; Moore, S.A.; Ford, G.C.; Campbell, K.P. Intracellular accumulation and reduced sarcolemmal expression of dysferlin in limb--girdle muscular dystrophies. Ann. Neurol. 2000, 48, 902–912.
- Selcen, D.; Stilling, G.; Engel, A.G. The earliest pathologic alterations in dysferlinopathy. Neurology 2001, 56, 1472–1481.
- McDade, J.R.; Michele, D.E. Membrane damage-induced vesicle-vesicle fusion of dysferlin-containing vesicles in muscle cells requires microtubules and kinesin. Hum. Mol. Genet. 2014, 23, 1677–1686.
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038.
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife 2013, 2, e00926.
- Draeger, A.; Babiychuk, E.B. Ceramide in plasma membrane repair. Handb. Exp. Pharmacol. 2013, 216, 341–353.
- Borgonovo, B.; Cocucci, E.; Racchetti, G.; Podini, P.; Bachi, A.; Meldolesi, J. Regulated exocytosis: A novel, widely expressed system. Nat. Cell Biol. 2002, 4, 955–962.
- Klinge, L.; Laval, S.; Keers, S.; Haldane, F.; Straub, V.; Barresi, R.; Bushby, K. From T-tubule to sarcolemma: Damage-induced dysferlin translocation in early myogenesis. FASEB J. 2007, 21, 1768–1776.
- Lek, A.; Evesson, F.J.; Lemckert, F.A.; Redpath, G.M.I.; Lueders, A.-K.; Turnbull, L.; Whitchurch, C.B.; North, K.N.; Cooper, S.T. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J. Neurosci. 2013, 33, 5085–5094.
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012, 26, 1875–1883.
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623.
- Waddell, L.B.; Lemckert, F.A.; Zheng, X.F.; Tran, J.; Evesson, F.J.; Hawkes, J.M.; Lek, A.; Street, N.E.; Lin, P.; Clarke, N.F.; et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 2011, 70, 302–313.
- Klinge, L.; Harris, J.; Sewry, C.; Charlton, R.; Anderson, L.; Laval, S.; Chiu, Y.-H.; Hornsey, M.; Straub, V.; Barresi, R.; et al. Dysferlin associates with the developing T-tubule system in rodent and human skeletal muscle. Muscle Nerve 2010, 41, 166–173.
- Kerr, J.P.; Ziman, A.P.; Mueller, A.L.; Muriel, J.M.; Kleinhans-Welte, E.; Gumerson, J.D.; Vogel, S.S.; Ward, C.W.; Roche, J.A.; Bloch, R.J. Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc. Natl. Acad. Sci. USA 2013, 110, 20831–20836.
- Demonbreun, A.R.; Rossi, A.E.; Alvarez, M.G.; Swanson, K.E.; Deveaux, H.K.; Earley, J.U.; Hadhazy, M.; Vohra, R.; Walter, G.A.; Pytel, P.; et al. Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity. Am. J. Pathol. 2014, 184, 248–259.
- Lukyanenko, V.; Muriel, J.M.; Bloch, R.J. Coupling of excitation to Ca2+ release is modulated by dysferlin. J. Physiol. (Lond.) 2017, 595, 5191–5207.
- Ampong, B.N.; Imamura, M.; Matsumiya, T.; Yoshida, M.; Takeda, S. Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol. 2005, 24, 134–144.
- Galbiati, F.; Engelman, J.A.; Volonte, D.; Zhang, X.L.; Minetti, C.; Li, M.; Hou, H.; Kneitz, B.; Edelmann, W.; Lisanti, M.P. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J. Biol. Chem. 2001, 276, 21425–21433.
- Lee, E.; Marcucci, M.; Daniell, L.; Pypaert, M.; Weisz, O.A.; Ochoa, G.-C.; Farsad, K.; Wenk, M.R.; De Camilli, P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 2002, 297, 1193–1196.
- Humphrey, G.W.; Mekhedov, E.; Blank, P.S.; de Morree, A.; Pekkurnaz, G.; Nagaraju, K.; Zimmerberg, J. GREG cells, a dysferlin-deficient myogenic mouse cell line. Exp. Cell Res. 2012, 318, 127–135.
- Philippi, S.; Bigot, A.; Marg, A.; Mouly, V.; Spuler, S.; Zacharias, U. Dysferlin-deficient immortalized human myoblasts and myotubes as a useful tool to study dysferlinopathy. PLoS Curr. 2012, 4, RRN1298.
- Mitchell, C.A.; McGeachie, J.K.; Grounds, M.D. Cellular differences in the regeneration of murine skeletal muscle: A quantitative histological study in SJL/J and BALB/c mice. Cell Tissue Res. 1992, 269, 159–166.
- Maley, M.A.; Fan, Y.; Beilharz, M.W.; Grounds, M.D. Intrinsic differences in MyoD and myogenin expression between primary cultures of SJL/J and BALB/C skeletal muscle. Exp. Cell Res. 1994, 211, 99–107.
- Ishiba, R.; Santos, A.L.F.; Almeida, C.F.; Caires, L.C.; Ribeiro, A.F.; Ayub-Guerrieri, D.; Fernandes, S.A.; Souza, L.S.; Vainzof, M. Faster regeneration associated to high expression of Fam65b and Hdac6 in dysferlin-deficient mouse. J. Mol. Histol. 2019, 50, 375–387.
- de Luna, N.; Gallardo, E.; Soriano, M.; Dominguez-Perles, R.; de la Torre, C.; Rojas-García, R.; García-Verdugo, J.M.; Illa, I. Absence of dysferlin alters myogenin expression and delays human muscle differentiation “in vitro”. J. Biol. Chem. 2006, 281, 17092–17098.
- Cohen, T.V.; Cohen, J.E.; Partridge, T.A. Myogenesis in dysferlin-deficient myoblasts is inhibited by an intrinsic inflammatory response. Neuromuscul. Disord. 2012, 22, 648–658.
- Belanto, J.J.; Diaz-Perez, S.V.; Magyar, C.E.; Maxwell, M.M.; Yilmaz, Y.; Topp, K.; Boso, G.; Jamieson, C.H.; Cacalano, N.A.; Jamieson, C.A.M. Dexamethasone induces dysferlin in myoblasts and enhances their myogenic differentiation. Neuromuscul. Disord. 2010, 20, 111–121.
- Zhang, Q.; Vashisht, A.A.; O’Rourke, J.; Corbel, S.Y.; Moran, R.; Romero, A.; Miraglia, L.; Zhang, J.; Durrant, E.; Schmedt, C.; et al. The microprotein Minion controls cell fusion and muscle formation. Nat. Commun. 2017, 8, 15664.
- Demonbreun, A.R.; Fahrenbach, J.P.; Deveaux, K.; Earley, J.U.; Pytel, P.; McNally, E.M. Impaired muscle growth and response to insulin-like growth factor 1 in dysferlin-mediated muscular dystrophy. Hum. Mol. Genet. 2011, 20, 779–789.
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell. Mol. Life Sci. 2013, 70, 4117–4130.
- Chiu, Y.-H.; Hornsey, M.A.; Klinge, L.; Jørgensen, L.H.; Laval, S.H.; Charlton, R.; Barresi, R.; Straub, V.; Lochmüller, H.; Bushby, K. Attenuated muscle regeneration is a key factor in dysferlin-deficient muscular dystrophy. Hum. Mol. Genet. 2009, 18, 1976–1989.
- Blau, H.M.; Cosgrove, B.D.; Ho, A.T.V. The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 2015, 21, 854–862.
- Doherty, K.R.; Cave, A.; Davis, D.B.; Delmonte, A.J.; Posey, A.; Earley, J.U.; Hadhazy, M.; McNally, E.M. Normal myoblast fusion requires myoferlin. Development 2005, 132, 5565–5575.
- Demonbreun, A.R.; Posey, A.D.; Heretis, K.; Swaggart, K.A.; Earley, J.U.; Pytel, P.; McNally, E.M. Myoferlin is required for insulin-like growth factor response and muscle growth. FASEB J. 2010, 24, 1284–1295.
- Posey, A.D.; Swanson, K.E.; Alvarez, M.G.; Krishnan, S.; Earley, J.U.; Band, H.; Pytel, P.; McNally, E.M.; Demonbreun, A.R. EHD1 mediates vesicle trafficking required for normal muscle growth and transverse tubule development. Dev. Biol. 2014, 387, 179–190.
- Davis, D.B.; Delmonte, A.J.; Ly, C.T.; McNally, E.M. Myoferlin, a candidate gene and potential modifier of muscular dystrophy. Hum. Mol. Genet. 2000, 9, 217–226.
- Yadav, A.; Kumar, B.; Lang, J.C.; Teknos, T.N.; Kumar, P. A muscle-specific protein “myoferlin” modulates IL-6/STAT3 signaling by chaperoning activated STAT3 to nucleus. Oncogene 2017, 36, 6374–6382.
- Posey, A.D.; Pytel, P.; Gardikiotes, K.; Demonbreun, A.R.; Rainey, M.; George, M.; Band, H.; McNally, E.M. Endocytic recycling proteins EHD1 and EHD2 interact with fer-1-like-5 (Fer1L5) and mediate myoblast fusion. J. Biol. Chem. 2011, 286, 7379–7388.
- Doherty, K.R.; Demonbreun, A.R.; Wallace, G.Q.; Cave, A.; Posey, A.D.; Heretis, K.; Pytel, P.; McNally, E.M. The endocytic recycling protein EHD2 interacts with myoferlin to regulate myoblast fusion. J. Biol. Chem. 2008, 283, 20252–20260.
- Lenhart, K.C.; O’Neill, T.J.; Cheng, Z.; Dee, R.; Demonbreun, A.R.; Li, J.; Xiao, X.; McNally, E.M.; Mack, C.P.; Taylor, J.M. GRAF1 deficiency blunts sarcolemmal injury repair and exacerbates cardiac and skeletal muscle pathology in dystrophin-deficient mice. Skelet Muscle 2015, 5, 27.
- Lenhart, K.C.; Becherer, A.L.; Li, J.; Xiao, X.; McNally, E.M.; Mack, C.P.; Taylor, J.M. GRAF1 promotes ferlin-dependent myoblast fusion. Dev. Biol. 2014, 393, 298–311.
- Melo, A.A.; Hegde, B.G.; Shah, C.; Larsson, E.; Isas, J.M.; Kunz, S.; Lundmark, R.; Langen, R.; Daumke, O. Structural insights into the activation mechanism of dynamin-like EHD ATPases. Proc. Natl. Acad. Sci. USA 2017, 114, 5629–5634.
- Leung, C.; Yu, C.; Lin, M.I.; Tognon, C.; Bernatchez, P. Expression of myoferlin in human and murine carcinoma tumors: Role in membrane repair, cell proliferation, and tumorigenesis. Am. J. Pathol. 2013, 182, 1900–1909.
- Aoki, M. Dysferlinopathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2015.
- Weiler, T.; Bashir, R.; Anderson, L.V.; Davison, K.; Moss, J.A.; Britton, S.; Nylen, E.; Keers, S.; Vafiadaki, E.; Greenberg, C.R.; et al. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s). Hum. Mol. Genet. 1999, 8, 871–877.
- Illarioshkin, S.N.; Ivanova-Smolenskaya, I.A.; Greenberg, C.R.; Nylen, E.; Sukhorukov, V.S.; Poleshchuk, V.V.; Markova, E.D.; Wrogemann, K. Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology 2000, 55, 1931–1933.
- Nakagawa, M.; Matsuzaki, T.; Suehara, M.; Kanzato, N.; Takashima, H.; Higuchi, I.; Matsumura, T.; Goto, K.; Arahata, K.; Osame, M. Phenotypic variation in a large Japanese family with Miyoshi myopathy with nonsense mutation in exon 19 of dysferlin gene. J. Neurol. Sci. 2001, 184, 15–19.
- Urtizberea, J.A.; Bassez, G.; Leturcq, F.; Nguyen, K.; Krahn, M.; Levy, N. Dysferlinopathies. Neurol. India 2008, 56, 289–297.
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017, 55, 55–68.
- Fanin, M.; Angelini, C. Progress and challenges in diagnosis of dysferlinopathy. Muscle Nerve 2016, 54, 821–835.
- Hornsey, M.A.; Laval, S.H.; Barresi, R.; Lochmüller, H.; Bushby, K. Muscular dystrophy in dysferlin-deficient mouse models. Neuromuscul. Disord. 2013, 23, 377–387.
- Sellers, S.L.; Milad, N.; White, Z.; Pascoe, C.; Chan, R.; Payne, G.W.; Seow, C.; Rossi, F.; Seidman, M.A.; Bernatchez, P. Increased nonHDL cholesterol levels cause muscle wasting and ambulatory dysfunction in the mouse model of LGMD2B. J. Lipid Res. 2018, 59, 261–272.
- Ho, M.; Post, C.M.; Donahue, L.R.; Lidov, H.G.W.; Bronson, R.T.; Goolsby, H.; Watkins, S.C.; Cox, G.A.; Brown, R.H. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum. Mol. Genet. 2004, 13, 1999–2010.
- Wenzel, K.; Geier, C.; Qadri, F.; Hubner, N.; Schulz, H.; Erdmann, B.; Gross, V.; Bauer, D.; Dechend, R.; Dietz, R.; et al. Dysfunction of dysferlin-deficient hearts. J. Mol. Med. 2007, 85, 1203–1214.
- Nishikawa, A.; Mori-Yoshimura, M.; Segawa, K.; Hayashi, Y.K.; Takahashi, T.; Saito, Y.; Nonaka, I.; Krahn, M.; Levy, N.; Shimizu, J.; et al. Respiratory and cardiac function in japanese patients with dysferlinopathy. Muscle Nerve 2016, 53, 394–401.
- Rosales, X.Q.; Moser, S.J.; Tran, T.; McCarthy, B.; Dunn, N.; Habib, P.; Simonetti, O.P.; Mendell, J.R.; Raman, S.V. Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. J. Cardiovasc. Magn. Reson. Off. J. Soc. Cardiovasc. Magn. Reson. 2011, 13, 39.
- Rubi, L.; Gawali, V.S.; Kubista, H.; Todt, H.; Hilber, K.; Koenig, X. Proper Voltage-Dependent Ion Channel Function in Dysferlin-Deficient Cardiomyocytes. Cell Physiol. Biochem. 2015, 36, 1049–1058.
- Chase, T.H.; Cox, G.A.; Burzenski, L.; Foreman, O.; Shultz, L.D. Dysferlin deficiency and the development of cardiomyopathy in a mouse model of limb-girdle muscular dystrophy 2B. Am. J. Pathol. 2009, 175, 2299–2308.
- Kitmitto, A.; Baudoin, F.; Cartwright, E.J. Cardiomyocyte damage control in heart failure and the role of the sarcolemma. J. Muscle Res. Cell. Motil. 2019, 40, 319–333.
- Han, R.; Bansal, D.; Miyake, K.; Muniz, V.P.; Weiss, R.M.; McNeil, P.L.; Campbell, K.P. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Invest. 2007, 117, 1805–1813.
- Tzeng, H.-P.; Evans, S.; Gao, F.; Chambers, K.; Topkara, V.K.; Sivasubramanian, N.; Barger, P.M.; Mann, D.L. Dysferlin mediates the cytoprotective effects of TRAF2 following myocardial ischemia reperfusion injury. J. Am. Heart Assoc. 2014, 3, e000662.
- Wei, B.; Wei, H.; Jin, J.-P. Dysferlin deficiency blunts β-adrenergic-dependent lusitropic function of mouse heart. J. Physiol. 2015, 593, 5127–5144.
- Lemckert, F.A.; Bournazos, A.; Eckert, D.M.; Kenzler, M.; Hawkes, J.M.; Butler, T.L.; Ceely, B.; North, K.N.; Winlaw, D.S.; Egan, J.R.; et al. Lack of MG53 in human heart precludes utility as a biomarker of myocardial injury or endogenous cardioprotective factor. Cardiovasc. Res. 2016, 110, 178–187.
- Wang, C.; Wong, J.; Fung, G.; Shi, J.; Deng, H.; Zhang, J.; Bernatchez, P.; Luo, H. Dysferlin deficiency confers increased susceptibility to coxsackievirus-induced cardiomyopathy. Cell. Microbiol. 2015, 17, 1423–1430.
- Shin, H.Y.; Jang, H.; Han, J.H.; Park, H.J.; Lee, J.H.; Kim, S.W.; Kim, S.M.; Park, Y.-E.; Kim, D.-S.; Bang, D.; et al. Targeted next-generation sequencing for the genetic diagnosis of dysferlinopathy. Neuromuscul. Disord. 2015, 25, 502–510.
- Sula, A.; Cole, A.R.; Yeats, C.; Orengo, C.; Keep, N.H. Crystal structures of the human Dysferlin inner DysF domain. BMC Struct. Biol. 2014, 14, 3.
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629.
- Evesson, F.J.; Peat, R.A.; Lek, A.; Brilot, F.; Lo, H.P.; Dale, R.C.; Parton, R.G.; North, K.N.; Cooper, S.T. Reduced plasma membrane expression of dysferlin mutants is attributed to accelerated endocytosis via a syntaxin-4-associated pathway. J. Biol. Chem. 2010, 285, 28529–28539.
- Voigt, T.; Sebald, H.-J.; Schoenauer, R.; Levano, S.; Girard, T.; Hoppeler, H.H.; Babiychuk, E.B.; Draeger, A. Annexin A1 is a biomarker of T-tubular repair in skeletal muscle of nonmyopathic patients undergoing statin therapy. FASEB J. 2013, 27, 2156–2164.
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Diaz Manera, J.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430.
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium. 2011, 50, 211–221.
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 2013, 58, 172–181.
- Kombairaju, P.; Kerr, J.P.; Roche, J.A.; Pratt, S.J.P.; Lovering, R.M.; Sussan, T.E.; Kim, J.-H.; Shi, G.; Biswal, S.; Ward, C.W. Genetic silencing of Nrf2 enhances X-ROS in dysferlin-deficient muscle. Front. Physiol. 2014, 5, 57.
- Beringer, A.; Gouriou, Y.; Lavocat, F.; Ovize, M.; Miossec, P. Blockade of Store-Operated Calcium Entry Reduces IL-17/TNF Cytokine-Induced Inflammatory Response in Human Myoblasts. Front. Immunol. 2018, 9, 3170.
- Nagaraju, K.; Rawat, R.; Veszelovszky, E.; Thapliyal, R.; Kesari, A.; Sparks, S.; Raben, N.; Plotz, P.; Hoffman, E.P. Dysferlin deficiency enhances monocyte phagocytosis: A model for the inflammatory onset of limb-girdle muscular dystrophy 2B. Am. J. Pathol. 2008, 172, 774–785.
- McNally, E.M.; Ly, C.T.; Rosenmann, H.; Mitrani Rosenbaum, S.; Jiang, W.; Anderson, L.V.; Soffer, D.; Argov, Z. Splicing mutation in dysferlin produces limb-girdle muscular dystrophy with inflammation. Am. J. Med. Genet. 2000, 91, 305–312.
- Yin, X.; Wang, Q.; Chen, T.; Niu, J.; Ban, R.; Liu, J.; Mao, Y.; Pu, C. CD4+ cells, macrophages, MHC-I and C5b-9 involve the pathogenesis of dysferlinopathy. Int. J. Clin. Exp. Pathol. 2015, 8, 3069–3075.
- Cárdenas, A.M.; González-Jamett, A.M.; Cea, L.A.; Bevilacqua, J.A.; Caviedes, P. Dysferlin function in skeletal muscle: Possible pathological mechanisms and therapeutical targets in dysferlinopathies. Exp. Neurol. 2016, 283, 246–254.
- Mariano, A.; Henning, A.; Han, R. Dysferlin-deficient muscular dystrophy and innate immune activation. FEBS J. 2013, 280, 4165–4176.
- Defour, A.; Medikayala, S.; Van der Meulen, J.H.; Hogarth, M.W.; Holdreith, N.; Malatras, A.; Duddy, W.; Boehler, J.; Nagaraju, K.; Jaiswal, J.K. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 2017, 26, 1979–1991.
- Wenzel, K.; Zabojszcza, J.; Carl, M.; Taubert, S.; Lass, A.; Harris, C.L.; Ho, M.; Schulz, H.; Hummel, O.; Hubner, N.; et al. Increased susceptibility to complement attack due to down-regulation of decay-accelerating factor/CD55 in dysferlin-deficient muscular dystrophy. J. Immunol. (Baltim. Md. 1950) 2005, 175, 6219–6225.
- Mitchell, C.A.; Grounds, M.D.; Papadimitriou, J.M. The genotype of bone marrow-derived inflammatory cells does not account for differences in skeletal muscle regeneration between SJL/J and BALB/c mice. Cell Tissue Res. 1995, 280, 407–413.
- Millay, D.P.; Maillet, M.; Roche, J.A.; Sargent, M.A.; McNally, E.M.; Bloch, R.J.; Molkentin, J.D. Genetic manipulation of dysferlin expression in skeletal muscle: Novel insights into muscular dystrophy. Am. J. Pathol. 2009, 175, 1817–1823.
- Roche, J.A.; Tulapurkar, M.E.; Mueller, A.L.; van Rooijen, N.; Hasday, J.D.; Lovering, R.M.; Bloch, R.J. Myofiber damage precedes macrophage infiltration after in vivo injury in dysferlin-deficient A/J mouse skeletal muscle. Am. J. Pathol. 2015, 185, 1686–1698.
- McElhanon, K.E.; Bhattacharya, S. Altered membrane integrity in the progression of muscle diseases. Life Sci. 2018, 192, 166–172.
- Lostal, W.; Bartoli, M.; Roudaut, C.; Bourg, N.; Krahn, M.; Pryadkina, M.; Borel, P.; Suel, L.; Roche, J.A.; Stockholm, D.; et al. Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy. PLoS ONE 2012, 7, e38036.

