Ferlins are multiple-C2-domain proteins involved in Ca2+-triggered membrane dynamics within the secretory, endocytic and lysosomal pathways. In bony vertebrates there are six ferlin genes encoding, in humans, dysferlin, otoferlin, myoferlin, Fer1L5 and 6 and the long noncoding RNA Fer1L4. Mutations in DYSF (dysferlin) can cause a range of muscle diseases with various clinical manifestations collectively known as dysferlinopathies, including limb-girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi myopathy. A mutation in MYOF (myoferlin) was linked to a muscular dystrophy accompanied by cardiomyopathy. Mutations in OTOF (otoferlin) can be the cause of nonsyndromic deafness DFNB9. Dysregulated expression of any human ferlin may be associated with development of cancer.
- dysferlin
- myoferlin
- otoferlin
- C2 domain
- calcium-sensor
- muscular dystrophy
- dysferlinopathy
- limb girdle muscular dystrophy type 2B (LGMD2B)
- membrane repair
- T-tubule system
- DFNB9
1. Introduction
2. Vertebrate Ferlins: Family Members and Domain Organization
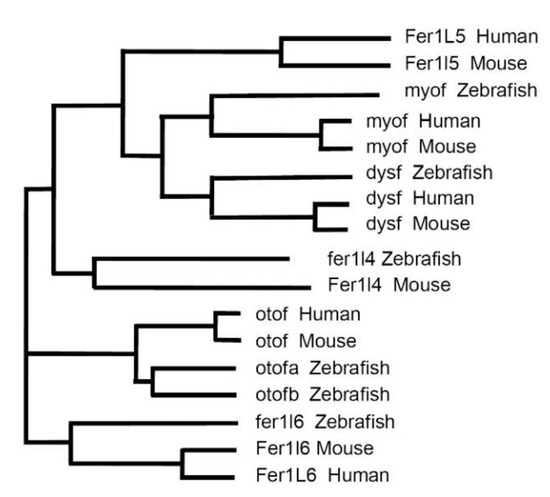
In the following discussion, we focused on the vertebrates—zebrafish (Danio rerio), mice (Mus musculus) and humans (Homo sapiens)—representing important (model) organisms for the study of ferlin functions. Six ferlin genes were present in each of these organisms (Figure 1). The phylogenetic analysis of the corresponding proteins demonstrating the evolutionary relationship between ferlins is shown in Figure 2.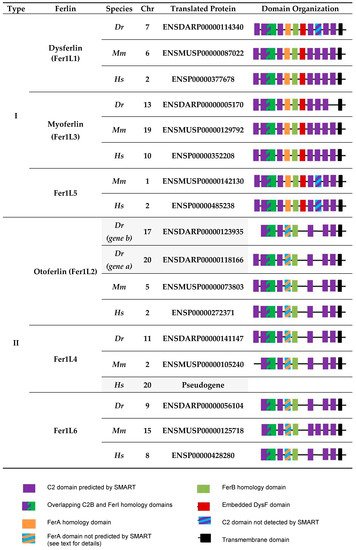
3. Functions of Dysferlin in Muscle
3.1. Dysferlin Functions in Sarcolemma Repair
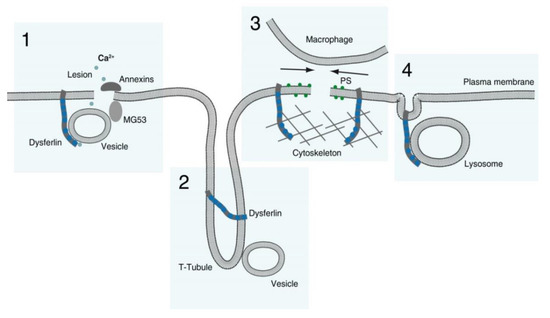
-
Membranous repair patch or plug formation;
-
T-tubule stabilization (see below) with T-tubule as a possible membrane reservoir;
-
PS-sorting; recruitment of macrophages and contraction of the membrane wound, and;
-
Lysosome exocytosis.
3.2. Dysferlin Functions in Triad Biology
3.3. Dysferlin in the Differentiation, Growth and Regeneration of Skeletal Muscle
4. Functions of Myoferlin and Fer1L5
5. Ferlins in Human Diseases: Dysferlinopathies and Their Pathomechanisms
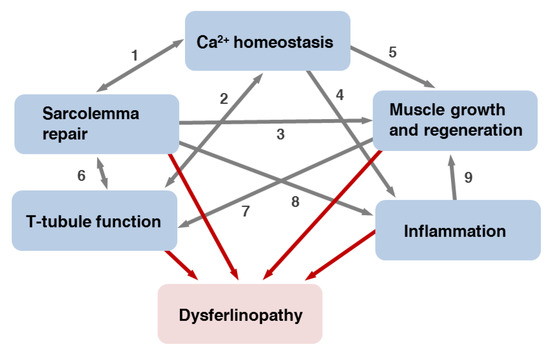
Dysferlinopathies are diseases caused by mutations in DYSF, affecting mainly skeletal muscles [80][180]. There are two common dysferlinopathy phenotypes—limb girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi myopathy (MM)—along with several more rare conditions: distal myopathy with anterior tibialis onset (distal anterior compartment myopathy), congenital muscular dystrophy and isolated hyperCKemia, an elevated concentration of serum creatine kinase (CK) [80][180]. Onset and progression of the disease as well as distribution of muscle weakness and wasting may vary significantly between individuals affected by dysferlinopathies. Several different clinical phenotypes can occur even within families carrying the same pathogenic variants of DYSF [5][81][82][83][5,181,182,183]. These observations emphasize the importance of investigating potential modifier genes [5][81][5,181]. Dysferlinopathies are characterized by late onset and slow progression. In carriers of pathogenic gene variants disease usually manifests in the second or third decade of life. The first symptoms are lower limb weakness accompanied by an increase in serum CK levels. The patients with the most severe phenotype of LGMD2B can become confined to wheelchair after two or three decades of disease progression, while most MM patients preserve ambulation [84][184]. Histological signs of the diseases are degeneration and regeneration of skeletal muscle [85][185] and in the more severe cases, fibrotic and adipogenic replacement of myofibers. On the protein level, dysferlinopathies are diagnosed by a complete loss or severe reduction of dysferlin in muscle biopsies or peripheral blood monocytes [86][186]. Mouse models lacking dysferlin develop muscular dystrophy, however, with an apparently less severe phenotype than humans and do not lose ambulation with age [87][88][187,188]. In dysferlin-deficient mice, the earliest symptoms are centrally nucleated fibers and marked differences in the myofiber diameter as well as 4- to 6-fold elevated CK levels at four weeks of age [89][189]. Latent cardiac dysfunction has been reported in dysferlinophathies, but patients do not primarily suffer from cardiomyopathies [90][91][190,191]. In a retrospective analysis, cardiac and respiratory functions were studied in dysferlinopathy patients [91][191]. Thus, a cardiovascular magnetic resonance analysis of LGMD2B patients showed indications of mild structural and functional cardiomyopathy [92][192]. One fifth of the patients developed respiratory problems and 9% required non-invasive ventilation. Accordingly, heart function in dysferlinopathy mouse models is either not or mildly affected [87][90][93][94][95][187,190,193,194,195]. However, also membrane repair in cardiomyocytes is dependent on dysferlin [96][196] and in a model of ischemia/reperfusion injury, dysferlin was shown to be cardioprotective [96][97][196,197]. Furthermore, physical stress exercise, or β-adrenergic activation provoke various symptoms of cardiac dysfunction in dysferlinopathy mice [90][94][96][97][98][99][190,194,196,197,198,199]. Mice with dysferlin inactivation show increased susceptibility to coxsackie virus infection and virus-induced myocardial damage [100][200], suggesting that pathways of viral infection and muscle repair may overlap. It is generally assumed that disease causing mutations are more or less uniformly distributed along the dysferlin-coding sequence [101][177] (Table 1). Two pathogenic missense mutations in human dysferlin FerA do destabilize the domain in differential scanning calorimetry (DSC) experiments [18][35] and a similar prediction was made for the three most frequent out of 15 missense mutations in the inner DysF domain [102][55]. These and other mutations may result in the poor dysferlin folding and degradation of the protein via different pathways. For example, missense mutation L1341P in C2E domain causes dysferlin aggregation in the ER and degradation by the additional autophagy/lysosome ER-associated degradation system [103][201]. Dysferlin lacking C2C domain or carrying patient mutation L344P within FerI domain demonstrate accelerated endocytosis, protein lability and endosomal proteolysis [104][47]. At present it is not clear how exactly loss-of-function mutations of DYSF and a decrease of the corresponding protein expression level lead to the development of dysferlinopathies. The following mechanisms may contribute to the development of the disease: (a) a defect in sarcolemma repair; (b) changes in Ca2+-homeostasis; (c) impaired muscle growth and regeneration and (d) inflammatory processes. Below, we discussed these factors in turn. To which degree, however, these mechanisms contribute to the development of the disease is still not known. The situation becomes even more complex when considering the existence of the various clinical manifestations of dysferlinopathies.5.1. Defective Repair of Myofiber Sarcolemma and, Possibly, T-Tubules
5.2. Changes in Muscle Fibers Ca2+ Homeostasis
5.3. Impaired Muscle Growth and Regeneration