 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sungjin Chung | + 1649 word(s) | 1649 | 2021-04-30 08:19:14 | | | |
| 2 | Rita Xu | Meta information modification | 1649 | 2021-05-13 06:24:21 | | |
Video Upload Options
The pharmacologic action of DPP-4 inhibitors is similar to that of GLP-1R agonists. The major therapeutic effects of DPP-4 inhibitors protect against degradation of the substrates GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), which are physiological substrates that affect insulin and glucagon secretion in a glucose-dependent manner.
1. Introduction
Chronic kidney disease (CKD) is a major public health burden, affecting more than 750 million people worldwide [1]. Because of the increasing global prevalence of type 2 diabetes mellitus (T2D) in CKD patients, CKD can be classified into diabetic kidney disease (DKD) and non-diabetic CKD. DKD accounted for 47% of patients initiating kidney replacement therapy due to end stage kidney disease (ESKD) in the United States in 2018 [2], and 48% in South Korea in 2019 [3].
During the past decade, a series of new anti-diabetic agents has been developed and validated to lower glycemia. Those drugs also carry cardiovascular and renal benefits and risks for patients with T2D. Thiazolidinediones can cause fluid retention and an increased risk of heart failure in patients with T2D [4]. However, glucagon-like peptide-1 receptor (GLP-1R) agonists are associated with favorable cardiovascular [5] and renal [6] outcomes in patients with T2D. Dipeptidyl peptidase-4 (DPP-4) inhibitors carry neither risk nor benefit to the cardiovascular system [7]. Sodium-glucose transporter-2 (SGLT-2) inhibitors emerged as game changers because they brought absolute benefits to cardiovascular [8] and renal [9] outcomes in patients with T2D.
The beneficial effects of these anti-diabetic agents on the cardiovascular system are independent of their glucose-lowering action. In particular, proteinuria reduction can be achieved by systemic or glomerular hemodynamic stability and inflammatory modulation. Consistent with that, GLP-1R agonists and SGLT-2 inhibitors reduce blood pressure and can preserve kidney function. As seen in the action of angiotensin II in the kidney and vasculature [10], hypertension, proteinuria, and renal inflammation are still the most important mediators for renal progression in both DKD and non-diabetic CKD. In contrast with DKD, no remarkable agents have been identified as effective measures to treat non-diabetic kidney disease during the past decade. Recent clinical trials elucidated the ability of SGLT-2 inhibitors to treat heart failure [11] and CKD [12] in patients without diabetes mellitus. However, clinical data are lacking to demonstrate their efficacy in specific non-diabetic kidney diseases.
2. Dipeptidyl Peptidase-4 Inhibitors
The pharmacologic action of DPP-4 inhibitors is similar to that of GLP-1R agonists. The major therapeutic effects of DPP-4 inhibitors protect against degradation of the substrates GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), which are physiological substrates that affect insulin and glucagon secretion in a glucose-dependent manner [13]. GLP-1 accumulates with the inhibition of DPP-4 because the soluble form of DPP-4 circulates in the plasma and rapidly degrades GLP-1. However, DPP-4 is also expressed as a membrane-bound form in a variety of tissues, primarily on endothelial and epithelial cells [14]. In the kidney, DPP-4 is expressed on the brush border of the proximal tubules and glomerular podocytes [15].
In addition to the extrapancreatic action derived from GLP-1, DPP-4 inhibitors can offer organ protection via GLP-1-independent mechanisms [16]. The enzyme DPP-4 cleaves multiple peptides other than GLP-1, such as brain-derived natriuretic peptide (BNP), neuropeptide Y (NPY), and stromal-derived factor (SDF)-1α. Thus, multiple substrates might be responsible for the pleiotropic action of DPP-4 inhibitors (Figure 1).
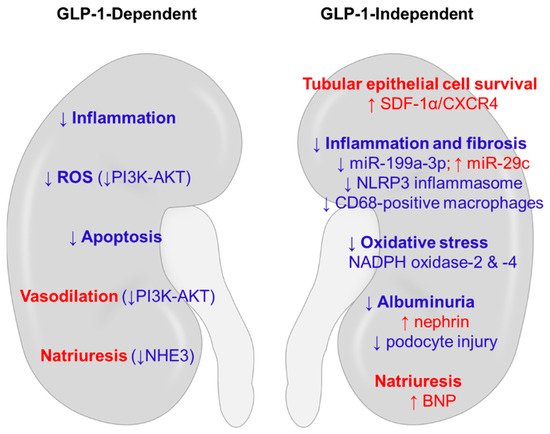
Figure 1. Potential mechanisms of renoprotection induced by DPP-4 inhibitors in non-diabetic kidney disease. The anti-inflammatory, anti-oxidative, and anti-apoptotic actions of DPP-4 inhibitors use both GLP-1-dependent and GLP-1-independent mechanisms. Hemodynamic benefits could be conferred through natriuresis and vasodilation. Red words denote stimulatory effects, and blue words denote inhibitory effects. BNP, brain-derived natriuretic peptide; CXCR4, C-X-C chemokine receptor type 4; DPP-4, dipeptidyl peptidase-4; GLP-1, glucagon-like peptide-1; NADPH, nicotinamide adenine dinucleotide phosphate; NHE3, Na+/H+ exchanger type 3; NLRP3, NLR family pyrin domain containing 3; PI3K-AKT, phosphatidylinositol 3-kinase-protein kinase B; SDF-1α, stromal-derived factor-1α.
2.1. Acute Kidney Injury
Vildagliptin pre-treatment in a rat model of ischemia/reperfusion injury preserved kidney function in association with reduced tubular necrosis and decreased apoptotic, oxidative, and proinflammatory markers [17]. Post-treatment with sitagliptin offered similar benefits in terms of kidney recovery and pleiotropic actions after acute ischemia/reperfusion injury [18].
Treatment with alogliptin reduced cisplatin–induced AKI and reduced the renal mRNA expression ratios of Bax/Bcl-2 and Bim/Bcl-2, markers of apoptosis [19]. In addition, the cisplatin-induced increase in the levels of other DPP-4 substrates, such as SDF-1α and NPY, was reversed. Teneligliptin also attenuated cisplatin-induced AKI and accelerated kidney recovery by promoting the proliferation of surviving epithelial cells in the proximal tubule via the chemokine ligand CXCL12 (or SDF-1α) and its receptor chemokine receptor 4 (CXCR4) [20]. Upregulation of the mRNA expression of both SDF-1α and CXCR4 was also found in the kidney after acute ischemia/reperfusion injury [21]. Thus, the SDF-1α/CXR4 axis could have a role in kidney repair by regenerating tubular epithelial cells in both ischemic and nephrotoxic injury. As shown in Figure 1, SDF-1α is an important DPP-4 substrate that potentially mediates the protective effects of DPP-4 inhibition in the kidney.
Natriuresis induced by DPP-4 inhibitors or GLP-1R agonists could be linked to renoprotection (Figure 1). Active sodium transport along the nephron is primarily driven by basolaterally located Na+-K+-ATPase that uses ATP hydrolysis as a source of energy. That process requires oxygen consumption to maintain a sustained rate of ATP generation in the kidney [22]. Na+/H+ exchanger type 3 (NHE3) is the major sodium transporter in the proximal tubule, and Girardi et al. reported that the administration of a DPP-4 inhibitor to Wistar rats for 7 days reduced both NHE3 activity and protein abundance in the proximal tubule brush border [23]. They also reported that NHE3 activity in LLC-PK1 cells was decreased by treatment with exendin-4 [24]. Downregulation of NHE3 could limit energy consumption in the proximal tubule and protect the kidney from acute ischemia/reperfusion injury [25].
2.2. Chronic Kidney Disease
As in diabetic nephropathy, albuminuria is an important marker of CKD and indicator of renal disease progression. Although DPP-4 inhibition appears to effectively ameliorate albuminuria [7], it is unlikely to improve renal survival in T2D patients [26]. It should be determined whether DPP-4 inhibitors are useful in patients with non-diabetic kidney disease.
Many preclinical studies have shown the renoprotective effects of DPP-4 inhibition in non-diabetic CKD. Alogliptin treatment ameliorated renal inflammation and fibrosis in mice with UUO [27]. Evogliptin also attenuated UUO-induced renal atrophy and tubulointerstitial fibrosis in association with the inhibition of pro-fibrotic gene expression and extracellular matrix production [28]. Consistent with that, linagliptin suppressed the induction of pro-fibrotic miRNA such as miR-199a-3p and restored levels of the anti-fibrotic miR-29c in rats with 5/6 nephrectomy [29]. Linagliptin also reduced albuminuria and attenuated glomerular hypertrophy and interstitial fibrosis in non-diabetic rats with 5/6 nephrectomy [30]. Joo et al. showed that in the rat remnant kidney model, sitagliptin improved renal functional and morphological changes by attenuating activation of the phosphatidylinositol 3-kinase (PI3K)-AKT pathway [31]. In aging mice, linagliptin improved kidney function and tubulointerstitial fibrosis in association with alterations to nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 and NADPH oxidase-4 [32].
The anti-inflammatory action of DPP-4 inhibition has also been shown in animal models of glomerulopathy. Alogliptin reduced the number of CD68-positive inflammatory macrophages in the kidney in a rat Thy-1 glomerulonephritis model [33]. Linagliptin pre-treatment in anti-GBM nephritic rats reduced the number of crescents, glomerulosclerosis, tubular injury, and renal fibrosis [34]. In mice with doxorubicin nephropathy, evogliptin reduced albuminuria in association with restored nephrin expression in podocytes and decreased podocyte injury [35]. Sitagliptin and linagliptin ameliorated NLRP3 inflammasome activation and oxidative stress markers in rats with doxorubicin nephropathy [36].
The anti-inflammatory action of DPP-4 inhibition was also demonstrated in animal models of salt-sensitive hypertension. Vildagliptin attenuated the development of salt-induced hypertension in Dahl salt-sensitive rats by increasing urine sodium excretion [37]. In addition, sitagliptin improved albuminuria and serum creatinine in Dahl salt-sensitive rats in association with the amelioration of inflammatory markers in the kidney [38]. Saxagliptin also improved albuminuria and suppressed inflammation- and fibrosis-related genes in Dahl salt-sensitive rats [39]. Table 1 summarizes the results of DPP-4 inhibitor treatment in animal models of non-diabetic kidney disease.
Table 1. Animal studies using DPP-4 inhibitors to treat non-diabetic kidney disease.
| Animal Model | Renal Function | Kidney Biomarker | Reference |
|---|---|---|---|
| Ischemia/reperfusion AKI (Wistar-Han rats) |
↓Serum creatinine | ↓Tubular damage and inflammation ↓Apoptosis ↓Oxidative stress ↓CXCL10 mRNA |
[17] |
| Ischemia/reperfusion AKI (Sprague-Dawley rats) |
↓BUN ↓Serum creatinine ↓Proteinuria |
↓Tubular injury ↓Oxidative stress ↓Pro-inflammatory markers ↓Apoptosis |
[18] |
| Cisplatin-induced AKI (C57BL/6 mice) |
↓BUN ↓Serum creatinine |
↓ATN score ↓Oxidative stress ↓Apoptosis |
[19] |
| Cisplatin-induced AKI (Sprague-Dawley rats) |
↓BUN ↓Serum creatinine |
↓Tubular injury ↓Interstitial fibrosis ↓Inflammation ↓Apoptosis ↑Proliferation of PTECs |
[20] |
| UUO (C57BL/6J mice) |
↔BUN ↔Serum creatinine |
↓Interstitial fibrosis ↓Pro-inflammatory markers |
[27] |
| UUO (C57BL/6J mice) |
N/A | ↓Interstitial fibrosis ↓Pro-fibrotic gene expression |
[28] |
| 5/6 nephrectomy (Wistar rats) |
↓Albuminuria ↓Proteinuria |
↓Interstitial fibrosis ↓Glomerular hypertrophy ↓Inflammation ↓Lipid peroxidation |
[30] |
| 5/6 nephrectomy (Sprague-Dawley rats) |
↓BUN ↑Creatinine clearance |
↓Glomerulosclerosis ↓Tubulointerstitial injury ↓PI3K-AKT activity ↓JNK phosphorylation ↓Apoptosis ↓Macrophage infiltration |
[31] |
| Aging C57BL/6 mice | ↓Serum creatinine ↓Cystatin C |
↓Mesangial matrix ↓Interstitial fibrosis ↓Pro-inflammatory markers ↓Oxidative stress |
[32] |
| Thy-1 glomerulonephritis (Sprague-Dawley rats) |
↓Proteinuria | ↓Glomerular injury ↓Macrophage infiltration |
[33] |
| Anti-GBM nephritis (Wistar Kyoto rats) |
↓Proteinuria | ↓Glomerulosclerosis ↓Crescents ↓Tubular injury ↓Inflammation ↓Podocyte injury |
[34] |
| Adriamycin nephropathy (BALB/c mice) |
↓Proteinuria ↓Albuminuria |
↓Macrophage infiltration ↑Podocyte number ↓Inflammation ↓Interstitial fibrosis |
[35] |
| Doxorubicin nephropathy (Sprague-Dawley rats) |
↔Proteinuria ↔Serum creatinine |
↓Tubular injury ↓Interstitial fibrosis ↓Inflammatory cell infiltration ↓Oxidative stress |
[36] |
| Hypertensive nephropathy (Dahl salt-sensitive rats) |
↓Albuminuria ↓Serum creatinine |
↓Interstitial fibrosis ↓Pro-inflammatory markers ↓Endothelial dysfunction ↓Oxidative stress |
[38] |
| Hypertensive nephropathy (Dahl salt-sensitive rats) |
↓Serum creatinine ↓Proteinuria ↓Albuminuria |
↓Vascular injury ↓Pro-inflammatory gene expression ↓Pro-fibrotic gene expression |
[39] |
Note: AKI, acute kidney injury; ATN, acute tubular necrosis; CXCL10, C-X-C motif chemokine ligand 10; GBM, glomerular basement membrane; IRI, ischemia-reperfusion injury; JNK, c-Jun N-terminal kinase; N/A, not available; NHE3, sodium hydrogen exchanger type 3; PCR, protein to creatinine ratio; PI3K, phosphatidylinositol 3-kinase; PTECs, proximal tubular epithelial cells; UUO, unilateral ureteral obstruction; ↑, increase; ↓, decrease; ↔, no significant change.
Although a few potential risks associated with DPP-4 inhibitors have been reported with respect to effects in the immune system and risk of acute pancreatitis, there is a relative lack of unwanted off-target or adverse effects associated with the DPP-4 inhibitors that are used therapeutically [13].
References
- Crews, D.C.; Bello, A.K.; Saadi, G. For the World Kidney Day Steering Committee Burden, Access, and Disparities in Kidney Disease. Nephron 2019, 141, 219–226.
- 2020 Annual Data Report from the United States Renal Data System. Available online: (accessed on 21 April 2021).
- Lee, J.Y.; Jin, D.-C. Patient characteristics according to rehabilitation and employment status in Korean hemodialysis patients. Kidney Res. Clin. Pr. 2020, 39, 356–364.
- Komajda, M.; McMurray, J.J.; Beck-Nielsen, H.; Gomis, R.; Hanefeld, M.; Pocock, S.J.; Curtis, P.S.; Jones, N.P.; Home, P.D. Heart failure events with rosiglitazone in type 2 diabetes: Data from the RECORD clinical trial. Eur. Heart J. 2010, 31, 824–831.
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. REWIND Investigators. Dulaglutide and car-diovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130.
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Botros, F.T.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and renal outcomes in type 2 diabetes: An exploratory analysis of the REWIND randomised, placebo-controlled trial. Lancet 2019, 394, 131–138.
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. CARMELINA Investi-gators. Effect of linagliptin vs. placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: The CARMELINA Randomized Clinical Trial. JAMA 2019, 321, 69–79.
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.H.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128.
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334.
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin System in Kidney Physiology. Compr. Physiol. 2014, 4, 1201–1228.
- Kosiborod, M.N.; Jhund, P.S.; Docherty, K.F.; Diez, M.; Petrie, M.C.; Verma, S.; Nicolau, J.C.; Merkely, B.; Kitakaze, M.; DeMets, D.L.; et al. Effects of dapagliflozin on symptoms, function, and quality of life in patients with heart failure and reduced ejection fraction: Results from the DAPA-HF trial. Circulation 2020, 141, 90–99.
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. DAPA-CKD Trial Committees and Investigators. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020, 383, 1436–1446.
- Deacon, C.F. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 642–653.
- Lambeir, A.-M.; Durinx, C.; Scharpé, S.; De Meester, I. Dipeptidyl-Peptidase IV from Bench to Bedside: An Update on Structural Properties, Functions, and Clinical Aspects of the Enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294.
- Kettmann, U.; Humbel, B.; Holzhausen, H.-J. Ultrastructural localization of dipeptidylpeptidase IV in the glomerulum of the rat kidney. Acta Histochem. 1992, 92, 225–227.
- Tanaka, T.; Higashijima, Y.; Wada, T.; Nangaku, M. The potential for renoprotection with incretin-based drugs. Kidney Int. 2014, 86, 701–711.
- Glorie, L.L.F.; Verhulst, A.; Matheeussen, V.; Baerts, L.; Magielse, J.; Hermans, N.; D’Haese, P.C.; De Meester, I.; De Beuf, A. DPP4 inhibition improves functional outcome after renal ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2012, 303, F681–F688.
- Chen, Y.T.; Tsai, T.H.; Yang, C.C.; Sun, C.K.; Chang, L.T.; Chen, H.H.; Chang, C.L.; Sung, P.H.; Zhen, Y.Y.; Leu, S.; et al. Exendin-4 and sitagliptin protect kidney from ischemia-reperfusion injury through suppressing oxidative stress and in-flammatory reaction. J. Transl. Med. 2013, 11, 270.
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Okamoto, K.; Negishi, K.; Nangaku, M.; Noiri, E. Protection of glucagon-like peptide-1 in cispla-tin-induced renal injury elucidates gut-kidney connection. J. Am. Soc. Nephrol 2013, 24, 2034–2043.
- Iwakura, T.; Zhao, Z.; Marschner, J.A.; Devarapu, S.K.; Yasuda, H.; Anders, H.J. Dipeptidyl peptidase-4 inhibitor teneligliptin accel-erates recovery from cisplatin-induced acute kidney injury by attenuating inflammation and promoting tubular regeneration. Nephrol Dial. Transpl. 2019, 34, 1669–1680.
- Tögel, F.; Isaac, J.; Hu, Z.; Weiss, K.; Westenfelder, C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005, 67, 1772–1784.
- Kiil, F.; Aukland, K.; Refsum, H.E. Renal sodium transport and oxygen consumption. Am. J. Physiol. Content 1961, 201, 511–516.
- Girardi, A.C.; Fukuda, L.E.; Rossoni, L.V.; Malnic, G.; Rebouças, N.A. Dipeptidyl peptidase IV inhibition downregulates Na+ -H+ ex-changer NHE3 in rat renal proximal tubule. Am. J. Physiol. Renal Physiol. 2008, 294, F414–F422.
- Carraro-Lacroix, L.R.; Malnic, G.; Girardi, A.C.C. Regulation of Na+/H+ exchanger NHE3 by glucagon-like peptide 1 receptor agonist exendin-4 in renal proximal tubule cells. Am. J. Physiol. Physiol. 2009, 297, F1647–F1655.
- Coppolino, G.; Leporini, C.; Rivoli, L.; Ursini, F.; di Paola, E.D.; Cernaro, V.; Arturi, F.; Bolignano, D.; Russo, E.; De Sarro, G.; et al. Exploring the effects of DPP-4 inhibitors on the kidney from the bench to clinical trials. Pharm. Res. 2018, 129, 274–294.
- MARLINA-T2D: Efficacy, Safety & Modification of Albuminuria in Type 2 Diabetes Subjects with Renal Disease with LINAgliptin. Available online: (accessed on 21 April 2021).
- Uchida, T.; Oda, T.; Matsubara, H.; Watanabe, A.; Takechi, H.; Oshima, N.; Sakurai, Y.; Kumagai, H. Renoprotective effects of a di-peptidyl peptidase 4 inhibitor in a mouse model of progressive renal fibrosis. Ren Fail. 2017, 39, 340–349.
- Kim, M.-J.; Kim, N.-Y.; Jung, Y.-A.; Lee, S.; Jung, G.-S.; Kim, J.-G.; Lee, I.-K.; Lee, S.; Choi, Y.-K.; Park, K.-G. Evogliptin, a Dipeptidyl Peptidase-4 Inhibitor, Attenuates Renal Fibrosis Caused by Unilateral Ureteral Obstruction in Mice. Diabetes Metab. J. 2020, 44, 186.
- Delić, D.; Wiech, F.; Urquhart, R.; Gabrielyan, O.; Rieber, K.; Rolser, M.; Tsuprykov, O.; Hasan, A.A.; Krämer, B.K.; Baum, P.; et al. Linagliptin and telmisartan induced effects on renal and urinary exosomal miRNA expression in rats with 5/6 nephrectomy. Sci. Rep. 2020, 10, 3373.
- Tsuprykov, O.; Ando, R.; Reichetzeder, C.; von Websky, K.; Antonenko, V.; Sharkovska, Y.; Chaykovska, L.; Rahnenführer, J.; Hasan, A.A.; Tammen, H.; et al. The dipeptidyl peptidase inhibitor linagliptin and the angiotensin II receptor blocker telmisartan show renal benefit by different pathways in rats with 5/6 nephrectomy. Kidney Int. 2016, 89, 1049–1061.
- Joo, K.W.; Kim, S.; Ahn, S.-Y.; Chin, H.J.; Chae, D.-W.; Lee, J.; Han, J.S.; Na, K.Y. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in rat remnant kidney. BMC Nephrol. 2013, 14, 98.
- Ban, T.H.; Kim, E.N.; Kim, M.Y.; Lim, J.H.; Lee, J.H.; Kim, H.D.; Yoon, H.E.; Park, C.W.; Choi, B.S. Renoprotective effect of a dipeptidyl pep-tidase-4 inhibitor on aging mice. Aging Dis. 2020, 11, 588–602.
- Higashijima, Y.; Tanaka, T.; Yamaguchi, J.; Tanaka, S.; Nangaku, M. Anti-inflammatory role of DPP-4 inhibitors in a nondiabetic model of glomerular injury. Am. J. Physiol. Physiol. 2015, 308, F878–F887.
- Mayer, A.L.; Scheitacker, I.; Ebert, N.; Klein, T.; Amann, K.; Daniel, C. The dipeptidyl peptidase 4 inhibitor linagliptin ameliorates renal injury and accelerated resolution in a rat model of crescentic nephritis. Br. J. Pharm. 2021, 178, 878–895.
- Lee, J.E.; Kim, J.E.; Lee, M.H.; Song, H.K.; Ghee, J.Y.; Kang, Y.S.; Min, H.S.; Kim, H.W.; Cha, J.J.; Han, J.Y.; et al. DA-1229, a dipeptidyl peptidase IV inhibitor, protects against renal injury by preventing podocyte damage in an animal model of pro-gressive renal injury. Lab. Investing. 2016, 96, 547–560.
- Jo, C.H.; Kim, S.; Park, J.-S.; Kim, G.-H. Anti-Inflammatory Action of Sitagliptin and Linagliptin in Doxorubicin Nephropathy. Kidney Blood Press. Res. 2018, 43, 987–999.
- Sufiun, A.; Rafiq, K.; Fujisawa, Y.; Rahman, A.; Mori, H.; Nakano, D.; Kobori, H.; Ohmori, K.; Masaki, T.; Kohno, M.; et al. Effect of dipeptidyl peptidase-4 inhibition on circadian blood pressure during the development of salt-dependent hypertension in rats. Hypertens. Res. 2015, 38, 237–243.
- Cappetta, D.; Ciuffreda, L.P.; Cozzolino, A.; Esposito, G.; Scavone, C.; Sapio, L.; Naviglio, S.; D’Amario, D.; Crea, F.; Rossi, F.; et al. Dipeptidyl peptidase 4 inhibition ameliorates chronic kidney disease in a model of salt-dependent hy-pertension. Oxid. Med. Cell. Longev. 2019, 2019, 8912768.
- Uchii, M.; Kimoto, N.; Sakai, M.; Kitayama, T.; Kunori, S. Glucose-independent renoprotective mechanisms of the tissue dipeptidyl peptidase-4 inhibitor, saxagliptin, in Dahl salt-sensitive hypertensive rats. Eur. J. Pharm. 2016, 783, 56–63.


