 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fengkai Li | + 3016 word(s) | 3016 | 2021-05-06 11:42:21 | | | |
| 2 | Conner Chen | Meta information modification | 3016 | 2021-05-12 11:35:00 | | |
Video Upload Options
Cancer stem cells (CSCs) refer to a subpopulation of tumor cells that have abilities to self-renew, differentiate, and seed new tumors, they might be taking part in tumor-associated angiogenesis via trans-differentiation or forming the capillary-like vascular mimicry (VM) in the tumor microenvironment. CSC-associated tumor neovascularization partially contributes to the failure of cancer treatment. The study of CSCs transdifferentiating to endothelial cells or pericytes can provide a new insight in the understanding of tumor progression and relapse.
1. Background
Cancer is a serious disease that threatens human health all over the world. Cancer treatment options include chemotherapy, radiotherapy, surgery, and some newly developed Immunotherapies. However, the efficiency of these treatments strongly depends on multiple factors such as cancer staging, drug-resistance, and even the tumor-microenvironment. Blood vessels play a crucial role in tumor progression to supply the oxygen and nutrients [1]. Until Folkman’s studies provided the evidence that tumors depend on angiogenesis [2], tumors were thought to acquire their blood supply from preexisting blood vessels. However, unfortunately, drugs or compounds aimed to suppress the angiogenesis, such as Bevacizumab blocking vascular endothelial growth factor (VEGF) and VEGF inhibitor vandetanib, have not achieved the desired effects [3][4]. In recent years, a growing number of reports have described that cancer stem cells (CSCs) are involved in drug-resistance and relapse [5][6][7]. CSCs refer to a subpopulation of tumor cells that have abilities to self-renew, differentiate, and seed new tumors [8]. In this regard, CSCs exhibit self-renewal and multilineage differentiation abilities; they might be taking part in tumor-associated angiogenesis via trans-differentiation or forming the capillary-like vascular mimicry (VM) in the tumor microenvironment.
2. Vascular Mimicry
2.1. Molecular Determinants Regulating the Formation of Vascular Mimicry
Regarding the significance of VM in malignant tumor progression, researchers have made great efforts to explore the molecular mechanisms driving the aggressive tumor cells to display the endothelial-like phenotype. Several key molecules have been found to regulate the formation of VM. Seftor et al. have already demonstrated several important cellular and molecular determinants of aggressive melanoma VM, including VE-Cadherin and EphA2 [9]. VE-Cadherin is an adhesion molecule previously thought to be exclusively expressed by endothelial cells [10][11]. However, it was found to be exclusively highly expressed in the aggressive melanoma cells and undetectable in the non-aggressive melanoma cells, supporting a vasculogenic-like patterned network of aggressive melanoma cells in three-dimensional culture. No networks could form when VE-Cadherin was down-regulated [9]. EphA2 is a protein belonging to ephrin receptor subfamily of the protein-tyrosine kinase family. It was also found to be expressed exclusively in the aggressive melanoma cells. Down-regulation of EphA2 abrogated the ability of tumor cells to form patterned networks [9][12] (Hess et al., 2001; Seftor et al., 2002). Furthermore, VE-Cadherin and EphA2 act in a coordinated manner in the regulation of vascular signaling pathway of melanoma cells. VE-Cadherin could interact with EphA2 and, thus, mediate the plasma membrane localization and phosphorylation of EphA2 [13].
Other molecular targets were also reported to participate in VM formation including the matrix matalloproteinases (MMPs) and the focal adhesion kinase (FAK). The microarray gene chip analysis revealed a significant increase of ECM component in aggressive melanoma cells, including the MMP-1, MMP-2, MMP-9, MT1-MMP, and Laminin5. The Laminin5γ2 chain fragmentation by active MT1/MMP and MMP2 is an essential step for aggressive melanoma cells to engage in VM formation [14]. FAK signaling blockade also inhibited the VM formation. It is reported that EphA2 drives the cleavage of Laminin 5 via activation of FAK signaling, which subsequently leads to the activation of extracellular signal-regulated kinase 1 and 2 (ERK1/2) and finally induces the active MT1/MMP and MMP2 through the PI3K pathway [13][15] (Figure 1).
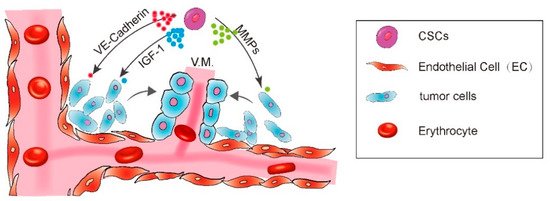
Figure 1. A diagram showing CSC enhance vasculogenic mimicry formation.
2.2. The Role of CSCs in Vascular Mimicry Formation
Besides the molecular determinants mentioned above, it is reported that the tumor cell VM is associated with stem cell characteristics. As early as the elaboration of VM by Maniotis et al., they compared the highly invasive versus poorly invasive melanoma tumor cells through cDNA microarray analysis, and found that the vascular mimetic invasive melanoma cells displayed a pluripotent embryonic-like genotype [16]. Furthermore, Seftor et al. found that vasculogenic mimetic melanoma cells expressed multiple genes that regulated cell plasticity in embryogenesis [9]. In glioblastoma, Ricci-Vitiani et al. found that a significant portion of the vascular endothelium shared the same genomic alteration as tumor cells in tumor xenografts produced from orthotopic or subcutaneous injection of glioblastoma stem-like cells (GSCs) in immunocompromised mice. The vascular endothelium contained a subset of tumorigenic cells that could generate highly vascularized tumors with VM. In addition, in vitro culture of GSCs in the endothelial conditions endowed them with an endothelial-like phenotype [17]. Moreover, the tumor cells lining VM channels were found to express the stem cell factor SOX2 and OCT4 in hepatocellular carcinoma [18], suggesting a new mechanism for CSC-mediated tumor VM formation. Recently, various studies have described the participation of CSCs in the formation of VM in multiple types of cancer. The presence of several CSC markers is associated with VM formation.
2.2.1. CD44+ CSCs
The cell surface protein CD44, a transmembrane receptor for hyaluronic acid, has been implicated in a diverse array of cellular function, including cell–cell and cell–matrix interaction, differentiation, proliferation, apoptosis, and motility [19][20]. In breast cancer, CD44+CD24− tumor cells were identified as tumorigenic cells. As few as 100 cells of this tumorigenic subpopulation were able to generate tumor in mouse xenograft model [21]. In microvascular endothelial cells, CD44 was demonstrated to play a key role in regulating the cell proliferation and survival via activating downstream Hippo signaling pathway and modulating the expression of CD31 and VE-Cadherin [22]. In addition, CD44 variant expressed on metastatic melanoma cells induced the VE-Cadherin phosphorylation at Y658 and Y731, and, thus, disrupted the endothelial junction assembly, finally promoted the melanoma transendothelial migration, documenting a critical role for CD44 in mediating vascular functions [23]. CD44 is significantly correlated with the VM presence in multiple solid tumors [24][25][26]. Recently, CD44 is also found to significantly correlate with VE-Cadherin expression and VM presence in oral squamous cell carcinoma [26]. In Ewing sarcoma tumors and breast carcinoma tumors, the CD44/c-Met signaling pathway was identified as the key regulator for VM through microarray analysis of aggressive cells (VM forming) and non-aggressive cells (non-VM forming). Both CD44 standard isoform and its splice variant CD44v6 were relevant to VM. Overexpression of CD44 in Ewing sarcoma tumor cells increased its interaction with extracellular matrix ligand hyaluronic acid and facilitated the formation of vasculogenic networks in vitro, whereas CD44 knockdown suppressed it [25], illuminating a critical role of CD44 in the process of VM formation.
2.2.2. ALDH+ CSCs
The enzyme aldehyde dehydrogenase 1 (ALDH1) has been shown to be a marker for both normal and malignant breast stem cells. Breast tumor cells with increased ALDH1 expression displayed the stem/progenitor properties and showed the strongest tumorigenic capacity in the mouse xenograft model [27]. Since then, ALDH has been proved to be a CSC marker in several solid tumors [28]. Both the ALDH1 expression and VM presence predicted poor disease-free survival and overall survival of breast cancer patients and colorectal cancer patients. The expression of ALDH1 was strongly associated with the presence of VM in breast cancer, especially in aggressive triple-negative breast cancer [29][30]. A recent study using the triple-negative breast cancer cell line HCC1937/p53 has further shown that the ALDH+ cells sorted by fluorescence-activated cell sorting markedly formed VM on Matrigel culture, whereas the ALDH− cells did not [31].
2.2.3. CD133+ CSCs
CD133, a pentaspan membrane glycoprotein, is one of the most well characterized markers of the CSCs in multiple types of cancer. Accumulating evidence has shown the importance of CD133 in regulating CSC-mediated tumorigenesis, metastasis, and chemoresistance [32]. CD133 is significantly correlated with VM presence in human renal cell carcinoma [24]. In acute leukemia, the adherent bone marrow stromal cells derived from CD133+/CD34+ stem cells were able to form the capillary-like structures on Matrigel through secreting the insulin growth factor-1 (IGF-1) [33]. CD133 was also demonstrated to be associated with VM phenotype in a study including 134 samples of breast cancer patients. The CD133+ subpopulation of MDA-MB-231 cell line expressed higher level of VM-related genes VE-Cadherin, MMP-2 and MMP-9, and was able to form the vasculogenic-like channels in the matrix culture [34]. The breast tumor cells lining VM channels on Matrigel were also found to express CD133. Via using a three-dimensional reconstructed image to show the spatial relationship between CSCs and VM, Sun et al. has provided direct evidence indicating that tumor cells lining VM channels were derived from CSCs [35].
Additionally, CSCs may provide more VM-related factors to synergize VM formation. For instance, the Nodal protein, a member of the transforming growth factor-β (TGF-β) superfamily, has been reported to be expressed by CSCs to maintain its stem cell-like properties, and promote the VM formation via Smad2/3 signaling pathway in the in vitro study [36][37]. In conclusion, CSCs facilitate the VM formation by promoting the VM-related gene expression and an endothelial-like phenotype, and probably lining VM channels directly. However, the CSC subpopulation is known to be heterogenous. Although several markers have been identified to isolate the CSC subgroups, they are not always completely overlapping. For example, Liu et al. has found that breast CSCs existed in at least two distinct states. Breast CSCs located in the invasive edge of tumors are characterized as CD44+CD24- and primarily quiescent, whereas breast CSCs located in the central region are more likely to express ALDH and more proliferative [38]. The heterogeneity of CSCs existed within tumors raises the questions that, which group of CSCs is most responsible for the VM formation, and what are the exact molecular mechanisms of regulating CSC-mediated VM formation. Moreover, the plastic transformation between different states of CSCs makes it much more complex. There is still a long way to go before we thoroughly understand the VM phenomenon.
2.3. Therapeutically Targeting Vascular Mimicry
As mentioned above, VM formation is an important aspect of the tumor neovascularization distinguished from the endothelial cell-dependent angiogenesis, playing crucial roles in tumor growth and metastasis. There have been several studies linking angiogenesis-promoting factors to VM. For example, VEGFA could induce the formation of VM in vitro via upregulating the expression of EphA2 and MMPs in ovarian cancer cells [39]. However, the upregulated expression of VEGF was not significant in VM-forming tumor cells compared with other VM-related genes [40]. What is more, the anti-angiogenic agents could not specifically target the vasculogenic CSCs population. Therefore, the therapeutic effects of clinical anti-angiogenic agents on VM are very limited. Both the anti-VEGFA monoclonal antibody Bevacizumab and the endostatin have been reported to have no effect on VM [41][42][43]. The application of Bevacizumab failed to block the vascular structures derived from CD133+ CSCs in glioblastoma [43]. In fact, Bevacizumab treatment even led to increased VM in tumors [44]. Accordingly, therapeutic inhibition of VM in combination with other anti-angiogenic therapies can inhibit the tumor vascularization to the utmost extent. Actually, there have been several drugs or compounds that show the VM inhibition effect. For example, application of the cytotoxic drug vincristine in combination with the tyrosine kinase inhibitor dasatinib could delete the VM channels via inhibition of VM-related indicators VE-Cadherin, FAK, PI3K, and MMPs [45]. The natural extract compounds such as Hinokitiol and brucine, also have shown an inhibition effect on VM channels [46][47]. However, currently there are still no specific therapies to inhibit VM formation. Since VM is closely correlated with and regulated by the CSC status, it is possible that therapies targeting the presence of CSCs in combination with the anti-angiogenic therapies may be the best strategy to eliminate the tumor vessel formation.
CSCs enhance the VM formation via IGF-1, VE-Cadherin, MMPs, and FAK pathways.
3. CSC-Derived Endothelial Cells
In general, VEGF plays a critical role in the formation of the embryonic circulatory system and growth of blood vessels from pre-existing vasculature [48]. The vasculogenesis relies on VEGF in normal tissues. Especially, in glioblastomas tissue, the glioma stem cells (GSCs) have been reported to promote angiogenesis via releasing high level of VEGF [49][50]. CSCs maintain the potential of transdifferentiating into various cell types. In recent decades, CSCs/tumor cell derived endothelial cells have been reported in a variety of solid tumors [17][51][52][53].
3.1. CSCs Transdifferentiate into Endothelial Cells
3.1.1. Glioblastoma
Glioblastoma is one of the most vascular-rich tumors. Ricci-Vitiani et al. reported that the CD31+/CD144+ endothelial cells from freshly dissociated glioblastoma specimens shared the same chromosomal alterations. Of endothelial cells, 20–90% contain the same chromosomal alterations as tumor cells in glioblastoma. Moreover, the authors observed that human-specific CD31+ protein were only expressed in the tissue that formed in subcutaneous xenografts generated by the injection freshly isolated CD133+/CD31− glioblastoma cells [17][54]. Similarly, the authors in another group employed GFP labeled glioblastoma mice model and successfully obtained GFP-positive tumor cells with CD31+ or CD34+ endothelial cell characteristics, particularly in the deep area of the lesions [53]. By mean of the flow cytometry, it is shown that 10–25% of endothelial cells (CD45−CD31+CD34+) were positive for GFP.
The phenomenon observed in those studies indicates that glioma stem cells or glioblastoma cells have the potential of differentiating into endothelial lineages.
3.1.2. Renal Carcinoma
Renal carcinoma is one kind of human malignancies, with high metastatic and poor prognosis. In the cause of study a new drug for renal carcinoma, the authors search for the presence of a tumor-initiating stem cell population in renal carcinomas [52], they separated the renal tumor cells and used the mesenchymal stem cell marker CD105 from human renal carcinomas. Afterwards, they found that CD105+ cells but not CD105− cells have the proficiency of differentiation into vWF/KDR/VEGFR3/CD31 positive endothelial cells in vitro; moreover, they observed the direct contribution to the presence of endothelial cells in tumor vessels at SCID mice xenograft. This is the proof that CD105+ renal CSCs can not only generate endothelial cells in vitro, but also give rise to vessels with a human origin in vivo.
3.1.3. Breast Cancer
It is well established that CD44+CD24−/low cells were recognized as breast cancer stem cells [21]. Bussolati et al. employed mammosphere culture to enrich the CD44+CD24− breast CSCs, and induced the CSC differentiation into endothelial cells that express several endothelial markers (e.g., CD31, VE-Cadherin, CD105 and vWF). They also showed that breast CSCs gave rise to endothelial cells in NOD/SCID mice xenograft [55]. Although this paper was short in explaining the mechanisms of how BCSC differentiation into endothelial cells, they highlighted the further research direction for breast CSC derived endothelial cells.
3.2. CSCs Transdifferentiate into Pericytes
Anatomically, blood vessels in tumors consist of endothelial cells and pericytes [51][56]. The pericytes play a pivotal role in maintenance of the blood–brain barrier, facilitation of vessel maturation, and initiation of vessel sprouting. Pericytes communicate with endothelial cells to regulate endothelial homeostasis [51][54][56][57]. Absence or disfunction of pericytes is correlated with increased metastasis in colorectal, prostate, pancreatic, and breast cancers [58]. Cheng et al. demonstrated that GSCs have the ability to transdifferentiate into pericytes [59]. The authors employed a lineage-specific fluorescence reporter system to validate that GSCs have the capacity to transdifferentiate into pericytes in vivo. By analyzing clinical samples, they found that the majority of tumor pericytes carry the same genetic alternations as neoplastic cells. Elimination of GSCs derived pericytes inhibited tumor growth. The authors further found the GSCs are recruited towards endothelial cells via SDF-1-CXCR4 axis, then the TGF-β induces GSCs give rise to pericytes (Figure 2).
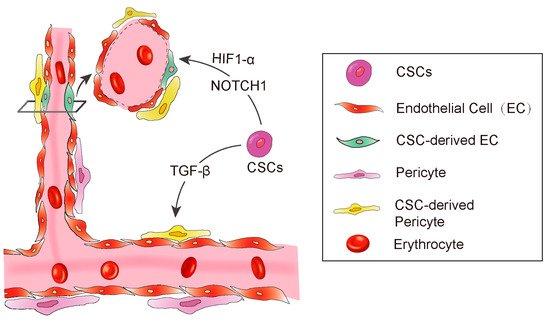
Figure 2. A diagram showing CSC derived angiogenesis. CSC-derived endothelial cells and pericytes play important roles in tumor vascular formation. Some signaling molecules, such as NOTCH1, HIF-1α, and TGF-β promote CSC to transdifferentiate into endothelial cells.
3.3. The Factors Affecting CSC Transdifferentiation
3.3.1. Hypoxia
Hypoxia inducible factor-1 (HIF-1α), a key transcriptional regulator for gene expression in response to hypoxia [60][61][62][63], is essential for hematopoietic stem cell to maintain the cell cycle quiescence [64]. Moreover, it has been reported to regulate angiogenesis via releasing some cytokines such as VEGF, platelet-derived growth factor (PDGF), and angiopoietin-1 (Ang-1) [62][63][65]. Soda et al. [53] employed the GFP-reporter mouse model and revealed a new way of VEGF-independent but Hif-1 induced GSC transdifferentiate into ECs. This study suggested that hypoxia, partly through the activation of HIF-1α, played an important role in the differentiation of glioblastoma stem cells to endothelial cells (Figure 2).
3.3.2. TGF-β
In addition to the essential roles in regulating differentiation, chemotaxis, proliferation, and activation of many immune cells, transforming growth factor-β (TGF-β) is also involved in maintaining cancer stemness and facilitating metastasis [66][67][68]. In Glioblastoma, TGF-β has been demonstrated to induce GSCs to transdifferentiate into pericytes [59]. They used Immunoblot to screen out the potential factors of inducing GSCs to transdifferentiate into pericytes, and found that the TGF-β greatly upregulates SAM expression in vitro. Furthermore, they also observed that coculture of GSCs and human brain microvascular endothelial cell line HBMEC enhanced the GSCs to transdifferentiate into pericytes and integrate into endothelial cells complexes, which were attenuated by the TGF-β antibody. In conclusion, these studies illustrated a clear route of GSC’s pericyte transdifferentiation: the endothelial cells recruit GSCs via the SDF-1/CXCR4 axis and then TGF-β induces GSC differentiation into pericytes. These studies strongly supported previous studies showing that TGF-β promotes breast cancer CSCs/early progenitor differentiation [69]. However, the source of TGF-β involved in GSC transdifferentiation was not demonstrated in these studies. Tumor cells could secrete TGF-β, and the immune cells that constitute the microenvironment could also secrete TGF-β. The identification of the source of TGF-β will provide more conveniences for the further research in the future (Figure 2).
3.3.3. NOTCH1
The NOTCH1 signaling pathway is important for cell–cell communication as well as it plays a key role in the development and regulation of angiogenesis [70]. Hovinga et al. showed that the CD133+ glioblastoma stem cells participate in endothelial hyperplasia and structure vascular glomeruloid bodies in a special 3D explant model. Further, they found that antagonised Notch results in a decrease in self-renewal potential of tumor CD133+ cells as well as a decrease in the number of cells or downregulation of CD133 expression in GSCs [71]. Another group has further demonstrated that NOTCH regulates angiogenesis in details [43]. They employed the NOTCH pathway inhibitor to treat CD133+/CD144− GSCs and observed that NOTCH inhibition resulted in significant suppression of the transition from CD133+/CD144− GSCs to CD133+/CD144+ endothelial progenitors (Figure 2).
All these studies demonstrate that the NOTCH1 pathway also regulates the GSC differentiation into endothelial cells in glioblastoma.
References
- Ribatti, D.; Vacca, A.; Dammacco, F. The Role of the Vascular Phase in Solid Tumor Growth: A Historical Review. Neoplasia 1999, 1, 293–302.
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186.
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803.
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887.
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284.
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751.
- Merlos-Suárez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Céspedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Muñoz, P.; et al. The Intestinal Stem Cell Signature Identifies Colorectal Cancer Stem Cells and Predicts Disease Relapse. Cell Stem Cell 2011, 8, 511–524.
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012.
- Seftor, E.A.; Meltzer, P.S.; Schatteman, G.C.; Gruman, L.M.; Hess, A.R.; Kirschmann, D.A.; Seftor, R.E.; Hendrix, M.J. Expression of multiple molecular phenotypes by aggressive melanoma tumor cells: Role in vasculogenic mimicry. Crit. Rev. Oncol./Hematol. 2002, 44, 17–27.
- Suzuki, S.; Sano, K.; Tanihara, H. Diversity of the cadherin family: Evidence for eight new cadherins in nervous tissue. Cell Regul. 1991, 2, 261–270.
- Lampugnani, M.-G.; Resnati, M.; Raiteri, M.; Pigott, R.; Pisacane, A.; Houen, G.; Ruco, L.; Dejana, E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J. Cell Biol. 1992, 118, 1511–1522.
- Hess, A.R.; Seftor, E.A.; Gardner, L.M.; Carles-Kinch, K.; Schneider, G.B.; Seftor, R.E.; Kinch, M.S.; Hendrix, M.J. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: Role of epithelial cell kinase (Eck/EphA2). Cancer Res. 2001, 61, 3250–3255.
- Hess, A.R.; Seftor, E.A.; Gruman, L.M.; Kinch, M.S.; Seftor, R.E.; Hendrix, M.J. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: Implications for vasculogenic mimicry. Cancer Biol. Ther. 2006, 5, 228–233.
- Seftor, R.E.B.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.G.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J.C. Cooperative Interactions of Laminin 5 γ2 Chain, Matrix Metalloproteinase-2, and Membrane Type-1-Matrix/Metalloproteinase Are Required for Mimicry of Embryonic Vasculogenesis by Aggressive Melanoma. Cancer Res. 2001, 61, 6322.
- Hess, A.R.; Postovit, L.-M.; Margaryan, N.V.; Seftor, E.A.; Schneider, G.B.; Seftor, R.E.; Nickoloff, B.J.; Hendrix, M.J. Focal adhesion kinase promotes the aggressive melanoma phenotype. Cancer Res. 2005, 65, 9851–9860.
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674.
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828.
- Zhao, X.; Sun, B.; Sun, D.; Liu, T.; Che, N.; Gu, Q.; Dong, X.; Li, R.; Liu, Y.; Li, J. Slug promotes hepatocellular cancer cell progression by increasing sox2 and nanog expression. Oncol. Rep. 2015, 33, 149–156.
- Ladeda, V.; Ghiso, J.A.A.; de Kier Joffé, E.B. Function and expression of CD44 during spreading, migration, and invasion of murine carcinoma cells. Exp. Cell Res. 1998, 242, 515–527.
- Hiraga, T.; Ito, S.; Nakamura, H. Cancer stem–like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013, 73, 4112–4122.
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988.
- Tsuneki, M.; Madri, J.A. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J. Biol. Chem. 2014, 289, 5357–5370.
- Zhang, P.; Fu, C.; Bai, H.; Song, E.; Song, Y. CD44 variant, but not standard CD44 isoforms, mediate disassembly of endothelial VE-cadherin junction on metastatic melanoma cells. FEBS Lett. 2014, 588, 4573–4582.
- Zhang, Y.; Sun, B.; Zhao, X.; Liu, Z.; Wang, X.; Yao, X.; Dong, X.; Chi, J. Clinical significances and prognostic value of cancer stem-like cells markers and vasculogenic mimicry in renal cell carcinoma. J. Surg. Oncol. 2013, 108, 414–419.
- Paulis, Y.W.; Huijbers, E.J.; van der Schaft, D.W.; Soetekouw, P.M.; Pauwels, P.; Tjan-Heijnen, V.C.; Griffioen, A.W. CD44 enhances tumor aggressiveness by promoting tumor cell plasticity. Oncotarget 2015, 6, 19634.
- Irani, S.; Dehghan, A. The expression and functional significance of vascular endothelial-cadherin, CD44, and vimentin in oral squamous cell carcinoma. J. Int. Soc. Prev. Community Dent. 2018, 8, 110.
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567.
- Toledo-Guzmán, M.E.; Hernandez, M.I.; Gómez-Gallegos, Á.A.; Ortiz-Sánchez, E. ALDH as a Stem Cell Marker in Solid Tumors. Curr. Stem Cell Res. Ther. 2019, 14, 375.
- Zhu, B.; Zhou, L.; Yu, L.; Wu, S.; Song, W.; Gong, X.; Wang, D. Evaluation of the correlation of vasculogenic mimicry, ALDH1, KAI1 and microvessel density in the prediction of metastasis and prognosis in colorectal carcinoma. BMC Surg. 2017, 17, 1–9.
- Xing, P.; Dong, H.; Liu, Q.; Zhao, T.; Yao, F.; Xu, Y.; Chen, B.; Zheng, X.; Wu, Y.; Jin, F. ALDH1 expression and vasculogenic mimicry are positively associated with poor prognosis in patients with breast cancer. Cell. Physiol. Biochem. 2018, 49, 961–970.
- Izawa, Y.; Kashii-Magaribuchi, K.; Yoshida, K.; Nosaka, M.; Tsuji, N.; Yamamoto, A.; Kuroyanagi, K.; Tono, K.; Tanihata, M.; Imanishi, M. Stem-like human breast cancer cells initiate vasculogenic mimicry on matrigel. Acta Histochem. Cytochem. 2018, 51, 173–183.
- Barzegar Behrooz, A.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2019, 27, 257–269.
- Mirshahi, P.; Rafii, A.; Vincent, L.; Berthaut, A.; Varin, R.; Kalantar, G.; Marzac, C.; Calandini, O.; Marie, J.; Soria, C. Vasculogenic mimicry of acute leukemic bone marrow stromal cells. Leukemia 2009, 23, 1039–1048.
- Liu, T.; Sun, B.; Zhao, X.; Zhao, X.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553.
- Sun, H.; Yao, N.; Cheng, S.; Li, L.; Liu, S.; Yang, Z.; Shang, G.; Zhang, D.; Yao, Z. Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer. Cancer Biol. Med. 2019, 16, 299.
- Gong, W.; Sun, B.; Zhao, X.; Zhang, D.; Sun, J.; Liu, T.; Gu, Q.; Dong, X.; Liu, F.; Wang, Y. Nodal signaling promotes vasculogenic mimicry formation in breast cancer via the Smad2/3 pathway. Oncotarget 2016, 7, 70152.
- Gong, W.; Sun, B.; Sun, H.; Zhao, X.; Zhang, D.; Liu, T.; Zhao, N.; Gu, Q.; Dong, X.; Liu, F. Nodal signaling activates the Smad2/3 pathway to regulate stem cell-like properties in breast cancer cells. Am. J. Cancer Res. 2017, 7, 503.
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91.
- Wang, J.Y.; Sun, T.; Zhao, X.L.; Zhang, S.W.; Zhang, D.F.; Gu, Q.; Wang, X.H.; Zhao, N.; Qie, S.; Sun, B.C. Functional significance of VEGF-a in human ovarian carcinoma: Role in vasculogenic mimicry. Cancer Biol. Ther. 2008, 7, 758–766.
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540.
- van der Schaft, D.W.; Seftor, R.E.; Seftor, E.A.; Hess, A.R.; Gruman, L.M.; Kirschmann, D.A.; Yokoyama, Y.; Griffioen, A.W.; Hendrix, M.J. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J. Natl. Cancer Inst. 2004, 96, 1473–1477.
- Liu, Z.; Li, Y.; Zhao, W.; Ma, Y.; Yang, X. Demonstration of vasculogenic mimicry in astrocytomas and effects of Endostar on U251 cells. Pathol. Res. Pract. 2011, 207, 645–651.
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833.
- Xu, Y.; Li, Q.; Li, X.-Y.; Yang, Q.-Y.; Xu, W.-W.; Liu, G.-L. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. 2012, 31, 16.
- Zeng, F.; Ju, R.-J.; Liu, L.; Xie, H.-J.; Mu, L.-M.; Zhao, Y.; Yan, Y.; Hu, Y.-J.; Wu, J.-S.; Lu, W.-L. Application of functional vincristine plus dasatinib liposomes to deletion of vasculogenic mimicry channels in triple-negative breast cancer. Oncotarget 2015, 6, 36625.
- Tu, D.G.; Yu, Y.; Lee, C.H.; Kuo, Y.L.; Lu, Y.C.; Tu, C.W.; Chang, W.W. Hinokitiol inhibits vasculogenic mimicry activity of breast cancer stem/progenitor cells through proteasome-mediated degradation of epidermal growth factor receptor. Oncol. Lett. 2016, 11, 2934–2940.
- Li, M.; Li, P.; Zhang, M.; Ma, F. Brucine suppresses breast cancer metastasis via inhibiting epithelial mesenchymal transition and matrix metalloproteinases expressions. Chin. J. Integr. Med. 2018, 24, 40–46.
- Karkkainen, M.J.; Petrova, T.V. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 2000, 19, 5598–5605.
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell–like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848.
- Liu, K.; Hao, M.; Ouyang, Y.; Zheng, J.; Chen, D. CD133+ cancer stem cells promoted by VEGF accelerate the recurrence of hepatocellular carcinoma. Sci. Rep. 2017, 7, 1–10.
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 2005, 7, 452–464.
- Bussolati, B.; Bruno, S.; Grange, C.; Ferrando, U.; Camussi, G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008, 22, 3696–3705.
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280.
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551.
- Bussolati, B.; Grange, C.; Sapino, A.; Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell. Mol. Med. 2009, 13, 309–319.
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427.
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood J. Am. Soc. Hematol. 2011, 118, 2906–2917.
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81.
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152.
- Guillemin, K.; Krasnow, M.A. The hypoxic response: Huffing and HIFing. Cell 1997, 89, 9–12.
- Wenger, R.H.; Gassmann, M. Oxygen (es) and the hypoxia-inducible factor-1. Biol. Chem. 1997, 378, 609–616.
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ. Res. 2003, 93, 664–673.
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55.
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402.
- Yoshida, D.; Kim, K.; Noha, M.; Teramoto, A. Hypoxia inducible factor 1-α regulates of platelet derived growth factor-B in human glioblastoma cells. J. Neuro-Oncol. 2006, 76, 13–21.
- Wang, M.-K.; Sun, H.-Q.; Xiang, Y.-C.; Jiang, F.; Su, Y.-P.; Zou, Z.-M. Different roles of TGF-β in the multi-lineage differentiation of stem cells. World J. Stem Cells 2012, 4, 28.
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769.
- Rao, S.; Zaidi, S.; Banerjee, J.; Jogunoori, W.; Sebastian, R.; Mishra, B.; Nguyen, B.N.; Wu, R.C.; White, J.; Deng, C. Transforming growth factor-β in liver cancer stem cells and regeneration. Hepatol. Commun. 2017, 1, 477–493.
- Tang, B.; Yoo, N.; Vu, M.; Mamura, M.; Nam, J.-S.; Ooshima, A.; Du, Z.; Desprez, P.-Y.; Anver, M.R.; Michalowska, A.M.; et al. Transforming growth factor-beta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 2007, 67, 8643–8652.
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689.
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029.


