 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sarmistha Saha | + 3527 word(s) | 3527 | 2021-04-30 13:24:32 | | | |
| 2 | Conner Chen | Meta information modification | 3527 | 2021-05-11 04:54:52 | | |
Video Upload Options
Wound healing involves a series of cellular events in damaged cells and tissues initiated with hemostasis and finally culminating with the formation of a fibrin clot. However, delay in the normal wound healing process during pathological conditions due to reactive oxygen species, inflammation and immune suppression at the wound site represents a medical challenge. So far, many therapeutic strategies have been developed to improve cellular homeostasis and chronic wounds in order to accelerate wound repair. In this context, the role of Nuclear factor erythroid 2-related factor 2 (Nrf2) during the wound healing process has been a stimulating research topic for therapeutic perspectives. Nrf2 is the main regulator of intracellular redox homeostasis. It increases cytoprotective gene expression and the antioxidant capacity of mammalian cells. It has been reported that some bioactive compounds attenuate cellular stress and thus accelerate cell proliferation, neovascularization and repair of damaged tissues by promoting Nrf2 activation.
1. Wound Healing Process
Wound healing begins immediately after wound formation and developed in three distinct phases, each of them associated with complex and diverse processes including hemostasis, inflammation, proliferation and maturation. In the hemostasis phase, the wound starts to be closed by clotting. Hemostasis begins just after blood leaks out of the body. At first, blood vessels constrict in order to restrict the blood flow. Then, the platelets coagulate to form a plug, while subsequently, a number of fibrin strands start to adhere and promote the formation of a thrombus, which entraps the blood cells in the wounded area. The second stage of healing is the inflammatory process, which starts immediately after blood vessels, and is characterized by an intense chemotaxis promoted by the leakage of a transudate that attracts the repair cells to the wounded site. This natural phase of the healing response is finalized to control bleeding and prevent infection by the removal of damaged cells and pathogens. In this stage, the typical signs of the inflammatory response known as heat, pain and redness, occur. While regarded as part of the physiological wound healing process, this phase can become detrimental if it prolongs or proceeds with excessive magnitude. In the proliferative phase, characterized by the formation of granulation tissue, new blood vessels and extensive re-epithelialization, the wound is rebuilt with extracellular matrix and collagen laid down by the fibroblasts, and then it begins to contract as a consequence of myofibroblasts activity. The granulation tissue is formed to provide sufficient oxygen and nutrients to achieve the optimal regeneration of the damaged tissue [1][2]. Afterwards, the re-epithelialization characterized by the intense proliferation and migration of keratinocytes towards the injury, progressively begins to resurface the lesion and concludes the proliferative phase of the wound process. The subsequent step, known as the maturation stage, is essentially a remodeling phase of the tissue wherein all the events activated in the early stages are markedly attenuated. In this stage, type III collagen is gradually replaced by type I collagen and the wound closes completely due to the crosslinking formation between the aligned collagen fibers. As a result, the scar thickness is progressively reduced whilst the tensile strength of the tissue is increased.
Given the complexity and the need for a sequential coordination of this multi-step process, failure in wound healing stages can lead to chronic wounds. Several factors can lead to impaired wound healing, including hypoxia, ischemia- reperfusion injury, bacterial colonization and altered collagen synthesis caused by systemic illness or chronic conditions including malnutrition and smoking, as well as local factors such as pressure, tissue edema and dehydration. Due to the imbalance between the production and degradation of collagen and growth factors, chronic wounds take a prolonged time to heal. Neutrophil infiltration to the wound site is regarded as an important factor for prolonged inflammation. Macrophages and neutrophils increase matrix metalloproteinase production, leading to extracellular matrix protein degradation [3].
Recent studies have shown that natural-based or synthetic compounds possess notable advantages in terms of wound healing process modulation via Nrf2 signaling as detailed in further sections. Many Nrf2 activators have been reported to control oxidative stress by regulating the redox homeostasis, therefore leading to improved wound healing [4].
2. Gene Structure of Nrf2 Transcription Factor and Its Repressor Keap1
Nrf2 is a basic leucine zipper (bZIP) transcription factor with Cap ‘n’ Collar (CNC) structure and interacts with the cysteine thiol groups of the protein Keap1 [5]. Keap1 acts as both an oxidative stress sensor and a regulator of the F-actin filament structure by virtue of its actin binding property. Nrf2 consists of seven functional Nrf2-ECh homology domains (Neh1-Neh7) [6]. The Neh1 domain provides the dimerization of Nrf2 with members of the musculoaponeurotic fibrosarcoma (Maf) protein and the DNA binding to the antioxidant response element (ARE) [7]. The Neh2 domain mediates the binding of Keap1 protein and Nrf2, and contains two different motifs, known as DLG and ETGE. The Neh3 domain is associated with stabilization of the protein, while the Neh4 domain is responsible for histone acetylation. The Neh4 and Neh5 domains are associated with transcriptional activation and promote the binding to the cAMP response element binding protein (CREBP). The Neh6 domain contains a number of serine residues that are recognized and subsequently phosphorylated by the protein glycogen synthase kinase 3 beta (GSK3β) leading to KEAP1-independent Nrf2 degradation. Lastly the Neh7 is an interaction domain with the nuclear receptor retinoic X receptor alpha (RXRa), that negatively controls Nrf2 activity. The Keap1 protein consists of five domains, an N-terminal region (NTR), a Tramtrack and Bric-à-Brac (BTB) domain, a central intervening region (IVR) with a nuclear export signal (NES) mediating the cytoplasmic localization of Keap1, six Kelch repeats and a C-terminal domain (CTR) [8]. The three domains, including, BTB, IVR and DGR domains, have binding sites. Highly reactive cysteine residues located within these domains (Cys151 and Cys226 in BTB, Cys273 and Cys288 in IVR) act as biochemical sensors of cellular stress and can be reversibly or irreversibly modified by ROS, RNS, H2S and other metabolites such as succinate or methylglyoxal. The DGR domain mediates the interaction between Keap1 and Nrf2 and directly binds to the ETGE motif of the Neh2 domain (Figure 1) [9][10].
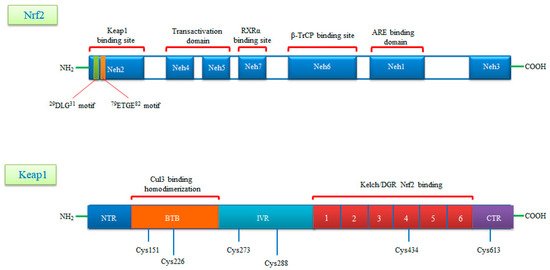
Figure 1. A schematic diagram of domain structures of Nrf2 and its repressor Keap1. Nrf2 consists of seven conserved domains known as Neh1-Neh7 and 605 amino acids. Neh1 interacts with small Maf (sMaf) proteins thanks to its Zip motif and thus binds to ARE sequences in DNA. The Neh2 domain contains two motifs called DLG and ETGE and interacts with the Keap1 molecule. The Neh4 and Neh5 domains are the CBP binding sites. Neh6 is a serine-rich domain that is required for TrCP binding and regulates Nrf2 stability. Neh7 is required to bind RXRα and inhibits ARE gene activity. Keap1, rich in cysteine residues, consists of five domains known as NTR, BTB, IVR, DGR, CTR and 624 amino acids. Keap1, rich in cysteine residues, consists of five domains known as NTR, BTB, IVR, DGR, CTR and 624 amino acids. The BTB domain provides homodimerization of Keap1 and is the binding site for Cullin3 (Cul3). The N-terminal region of the IVR domain mediates these functions and acts as a sensor for Nrf2 inducers. The Kelch/DGR domain inhibits the multiple ubiquitination of Nrf2 by mediating the binding of Nrf2 to the Neh2 domain.
3. Nrf2 Signaling: Mechanisms of Activation and Suppression
3.1. Canonical Activation of Nrf2
Under physiological conditions, Keap1 forms a homodimer that binds to the ETGE and DLG motifs in the Neh2 domain of Nrf2 causing its cytoplasmic retention. Keap1 is a substrate adaptor protein for the Cul3-based E3–ligase complex for ubiquitination and degradation by the ubiquitin proteosome system (UPS). It is estimated that under homeostatic conditions Nrf2 has a half-life of only 20 min, suggesting that its constant degradation is required to prevent unnecessary activation of the cytoprotective response [11][12][13].
By contrast, in the presence of cellular stress inducers such as ROS, xenobiotics and other electrophilic molecules, Nrf2 is unable to efficiently interact with the ubiquitin conjugating machinery due to a conformational change in the E3–ligase complex induced by the electrophilic modification of specific cysteines within Keap1 IVR or BTB domains [14]. Importantly, Keap1 is able to directly detect oxidative changes in the intracellular milieu, thanks to the presence of redox-sensitive cysteine residues located within its regulatory domains that are subject to covalent modifications when Keap1 is exposed to electrophilic Nrf2-inducing chemicals such as hydrogen peroxide [15]. As a consequence, the inactive Keap1 molecules are progressively saturated by the pre-existing pool of Nrf2 while the neosynthesized Nrf2 molecules can escape Keap1 negative regulation and enter into the nucleus. Of note, Nrf2 can be activated by a number of different ARE inducers. Among them, hydrogen peroxide (H2O2) [16], NO [17], tertiary butylhydroquinone (tBHQ) [18], dimethyl fumarate (DMF) [19] as well as some phytochemicals (Resveratrol, silymarin, sulforaphane, curcumin, cinnamic aldehyde etc.) [20] and bardoxolone methyl [21] are among the most well studied. They could protect the skin cells from UV damage and toxic chemicals. In addition, keratinocyte growth factor (KGF) and FGF, which are released by dermal fibroblasts after wound formation, also promote Nrf2 expression [22]. Heme released from heme proteins is required for Nrf2 stabilization in case of damage [23]. Additionally, Nrf2 can be activated through mitogen-activated protein kinase (MAPK) and protein kinase C (PKC) signaling pathways in response to oxidative stress [24].
After Nrf2 is released from Keap1 and translocated into the nucleus, it is dimerized with the sMaf protein and binds to the ARE. It further regulates the expression of antioxidant enzymes and genes encoding cell protective proteins such as NAD(P) H: quinone oxidoreductase (NQO)-1, glutathione reductase (GSR), glutathione-S transferase (GST), glutathione peroxidase (GPx) superoxide dismutases 1–3 (SOD1–3), glutamate-cysteine ligase catalytic subunit (GCLC), peroxiredoxins (PRX), thioredoxins (TRX), catalase (CAT), heme oxygenase-1 (HO-1) and many others (Figure 2) [12][25].
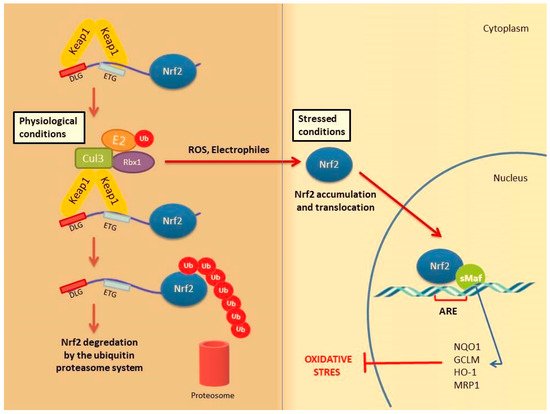
Figure 2. Nrf2 activation mechanism-canonical pathway. Under normal physiological conditions, Nrf2 forms a complex with the Keap1 protein Cul3 and Rbx, causing Nrf2 to be ubiquitinated and degraded by the ubiquitin proteasome system Under stress conditions, the Keap1 Cul3 complex is deactivated. This situation leads to the release and accumulation of Nrf2. Stable Nrf2 is translocated to the nucleus and binds to ARE together with small Maf proteins (sMaf). It ultimately activates antioxidant enzymes and cytoprotective proteins such as HO-1, NQO-1, GST. These enzymes support cellular defense by mediating the removal of ROS and cytotoxic electrophiles.
3.2. Non-Canonical Activation of Nrf2: The Role of Autophagy
A number of alternative regulators of Nrf2 activation have been described and are collectively ascribed in the non-canonical pathway of Nrf2 activation. This pathway comprises a number of different proteins such as DPP3, PALB2, BRCA1, p62 and p21, possessing the ability to disrupt the formation of the Keap1-Nrf2 complex by preventing their reciprocal interaction through competitive binding, thus promoting Nrf2 stabilization and activation [26]. Possibly, the most well-studied mechanism of the non-canonical pathway is linked to autophagy. Autophagy is a highly conserved process involving the reuse of long-lived proteins and building blocks that are released as a result of the breakdown of damaged organelles. This conserved mechanism contributes to the restoration of cellular homeostasis and is activated in response to specific conditions such as nutrient scarcity, ROS overproduction, genomic instability, misfolded protein accumulation, organelle damage and microbial infection. Accumulating evidence also suggests that the autophagy mechanism can be triggered due to excessive ROS in cells. This mechanism allows misfolded or damaged proteins and damaged organelles to be broken down and reused for the cell purposes. Apart from this, it is also a vital defense mechanism in protecting against proteotoxicity induced by cellular redox stress [27]. Both autophagy and Nrf2/Keap1 have a protective role against oxidative stress (Figure 3).
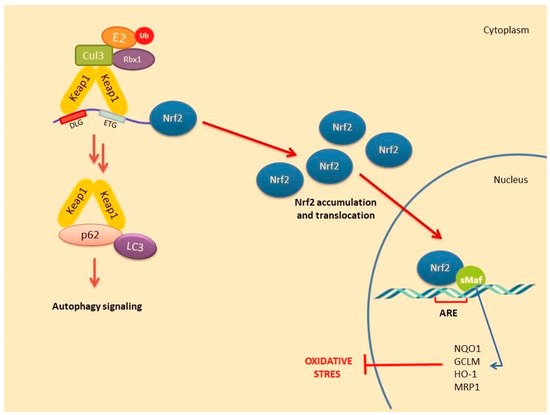
Figure 3. Nrf2 activation mechanism-canonical pathway associated with the induction of autophagy.
In this pathway, p62 protein, which is an adapter protein that binds to ubiquitinated protein aggregates in autophagy and transmits it to autophagosomes. Upon serine phosphorylation within its 349-DPSTGE-354 motif, the p62 protein interacts with 3 arginines in the Kelch domain of Keap1, thus preventing or destabilizing Nrf2 binding. This further leads to Nrf2 stabilization and subsequent nuclear translocation inducing the transcription of several target genes [28]. Downmodulation of the p62 levels can thus restore the negative regulation of Keap1 and decrease Nrf2 stability [29][30][31].
4. Nrf2 Activation during Wound Healing
The main role of Nrf2 in wound healing is to detect the ROS accumulation in injured and inflamed tissues and to activate the antioxidant defense system [32]. Therefore, pharmacological induction of Nrf2 is an important therapeutic target in promoting healing after tissue damage and controlling repair-related inflammation [33]. Nrf2 pathway plays a protective role against oxidative stress through the expression of antioxidative enzymes during wound healing [34][35].
High ROS levels inhibit the proliferation of chondrocytes, stimulate apoptosis of osteocytes and increase bone resorption causing bone loss or delayed healing. Nrf2 transcription factor is expressed in many cell types including osteoblasts, osteocytes and osteoclasts. Lippross et al. showed that Nrf2 deficiency impairs normal fracture healing in Nrf2 knockout (KO, Nrf2−/−) mice [36]. Similarly, in Nrf2-KO mice, the absence of Nrf2 suppresses the expression of antioxidant enzymes in osteoblasts [37]. Although these results suggest a curative role of Nrf2 pathway during fracture healing, more studies are needed to better understand Nrf2 function in different bone cells [33].
Many growth factors such as basic FGF and insulin growth factor-1 (IGF-1) are responsible for modulating cellular responses after bone injury mediate fracture healing. Vascular endothelial growth factor (VEGF), one of the most crucial ones, promotes increased oxygenation in the injury site and initiates neovascularization. On the other hand, VEGF triggers osteoblast proliferation and migration [38]. Kweider et al. showed that VEGF activates Nrf2 in the BeWo cell line and enhances the levels of some antioxidant enzymes (thioredoxin, thioredoxin reductase and HO-1) [39]. However, this activation depends on the upstream induction of the ERK1/2 signal. These results confirm that Nrf2 promotes fracture healing by inducing antioxidant responses to protect cells from damage caused by ROS in bone damage.
During hemolysis, toxic-free heme is released by red blood cells. The conversion of heme to carbon monoxide and free iron is catalyzed by HO-1, which has an important immunomodulatory role. Indeed, it has been reported to have an anti-inflammatory effect by increasing the release of cytokines such as interleukin 10 (IL 10) and IL1β. Inhibition of free heme release by hemolysis is triggered by HO-1 expression caused by Nrf2 activation. In K565 cells (human pro-erytroid cells), hemin causes the ubiquitination of Keap1 and promotes Nrf2 stabilization. Therefore, targeting of the Nrf2/Keap1 system has been proposed to prevent heme toxicity [40].
Several reports have suggested that skin damage after UVB irradiation can lead to DNA modification and ROS formation [41]. In these cases, pharmacological activation of Nrf2 could be an effective way of reducing UVB cytotoxicity. In transgenic mice, endogenous Nrf2 prevents ROS damage caused by UVB and apoptosis of keratinocytes by inducing the activation of cytoprotective genes [42]. Topical application of Nrf2 activators sulforaphane (SFN) has been shown to protect the skin against acute UV toxicity [5]. In the bleomycin (BLM)-induced skin fibrous model, BLM was found to inhibit Nrf2 expression in the epidermis. However, Nrf2 deficiency in keratinocytes exacerbated skin fibrosis. This deficiency could also be correlated with the increased expression levels of Mcp-1, IL-6 and IL-8 [43]. Additionally, the formulation named RTA408 was shown to increase the expression of Nrf2 target genes and mediate re-epithelization [44]. Based on these evidence, Nrf2 pathway has a remarkable impact on healing of wounds and may be beneficial in the treatment of chronic wounds. Selected studies targeting Nrf2 activation against tissue damage in different experimental models are given in Table 1.
5. The Role of Nrf2 in Cell Proliferation, Apoptosis and Migration during Wound Repair
Nrf2 activation stimulates the proliferation and migration of epithelial cells during wound repair and inhibits apoptosis [45]. Loss of Nrf2 further slowed the epithelization process in the STZ-induced diabetic mouse model. This suggests that the deficiency of Nrf2 also impairs angiogenesis due to long-term inflammation and lack of neovascularization mediators. This is caused by the concomitant effects of increased apoptosis and oxidative damage accompanied by low TGF and high MMP levels. Nrf2 activation can be triggered by MMP9, transforming growth factor-β (TGF-β) and genes associated with migration and proliferation that promote wound healing in perilesional skin tissue taken from diabetic and ulcer patients [46].
Endothelial dysfunction represents a further underlying deterrent to wound healing, especially in diabetes. A number of studies have suggested that bone-marrow derived endothelial progenitor cells (EPCs) contribute to postnatal neovascularization and vascular endothelial repair [47][48]. The number of circulating EPCs in humans [49] and in animals [50] with diabetes mellitus (DM) was reduced with a concomitant reduction in EPC functional ability (proliferation, colony formation, tube formation, self-renewal and mobilization) [51][52]. Recently, it has been reported that Nrf2 activation protects diabetic EPCs against the effects of oxidative stress and cell senescence, ameliorating the biological dysfunction of EPCs derived from mice with diabetes [53]. Thus, the targeting of endothelial dysfunction may represent another promising focus for future treatment of the diabetic wound with Nrf2 activators (Figure 4).
Table 1. Studies targeting Nrf2 activation against tissue damage.
| Model | Injury | Method of Application | Treatment | Studied on | Results | Ref |
|---|---|---|---|---|---|---|
| in vivo | BLM-induced skin fibrosis | s.c. injection | - | Nrf2−/− mice generated with Keratin 14-Cre/loxp system |
Increased cytokines & chemokines expression (Mep-1, IL-6, IL-8) | [43] |
| in vivo | Corneal epithelial injury | i.m. injection | - | Nrf2−/− mice | Induction of cell migration and inhibition of cell proliferation | [45] |
| in vivo | Endothelial cell injury | skin incision | - | Nrf2−/− C57BL/6 mice | VEGF-induced proliferation ↓ Endothelial cell sprout formation ↓ |
[54] |
| in vitro | Retinal pericytes, astrocytes and endothelial cells | - | - | Blood-retinal barrier model | Increased IL-1β, IL-6, iNOS, Nox2 expression Activation of Nrf2 and HO-1 |
[55] |
| in vivo | Epidermal keratinocyte | i.p. injection (dorsal skin 10 mm) |
- | Leprdb/db mice | Increased expression of Nqo1 and Sod2 genes due to impaired Nrf2 activity | [56] |
| in vivo | Nonhealing skin ulcers |
i.p. injection | - | Nrf2−/− C57BL/6 mice Perilesional skin tissue samples from diabetic patients |
Increasing proliferation and migration, decreasing apoptosis Upregulation of TGF-β1 and downregulation of MMP9 |
[57] |
| in vitro | Endothelial dysfunction | - | - | Primary human coronary arterial endothelial cells (CAECs) |
Impaired angiogenic processes (including proliferation, adhesion, migration and ability to form capillary-like structures) | [58] |
| in vivo | Chronic venous insufficiency-related wound injury |
i.p. injection | RTA 408 (Omaveloxolone) | C57BL/6 mice | Expression of antioxidant mediators | [59] |
| in vivo | Bed wounds | s.c. injection (dorsal skin 12 mm) |
ESC-Exos treatment | C57BL/6 mice skin aging model | Nrf2 activation, improved skin aging and downregulation of Keap1 by miR-200a | [60] |
| in vivo | Retina injury | intravitreally injection | MIND4-17 | BALB/C mice | Activation of Nrf2 and reduced disfunction of retina Preventing apoptosis caused by high glucose |
[61] |
| in vivo | Atherosclerotic lesions | i.p. injection | tBHQ | apoE−/− mice | Increased expression of HMOX1, SOD1 and CAT Upregulation of autophagy-related genes (BECN1, SQSTM1/p2, ATG5/7) |
[62] |
| in vivo | Impaired wound healing | i.p. injection | DMF | Wistar rats | Upregulation of Nqo1 and HO-1 expression Downregulation of IL1β, IL-6 and MCP1 |
[63] |
| in vivo | Impaired wound healing | i.p. injection (1.5 mm dorsal skin) |
LPS-Exos (500 µg/mL –1 mg/mL, 21 day) |
Sprague Dawley rats | Increased expression of Nrf2, HO-1 and Nqo1 genes Decreased expression of proinflammatory cytokines (IL-6, IL-1β, TNF-α) and MMP9 |
[64] |
| in vivo | Impaired wound healing | i.p. injection (8 mm dorsal skin) | PCB2 treatment (10 mg/kg daily) |
C57BL/6 mice | Promoting cell survival and migration Decreased oxidative stress | [65] |
| in vitro in vivo |
Cutaneous wound | i.p. injection (10 mm dorsal skin) |
siKeap1 | 3T3 cells (added siRNA-liposomal complex) Leprdb/db mice |
Redox homeostasis | [66] |
i.m.: intramuscular; i.p.: intraperitoneal; s.c.: subcutaneous.
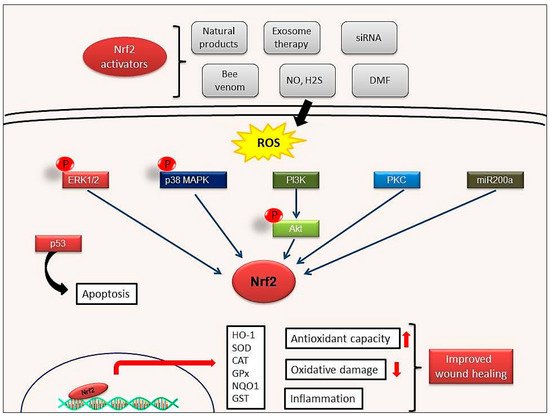
Figure 4. Possible wound healing mechanisms of Nrf2 activation. Activation of ERK1/2, p38MAPK, PI3K, PKC signaling pathways by Nrf2 activators. ERK1/2: Extracellular signal-regulated protein kinases 1 and 2; p38MAPK: p38 mitogen-activated protein kinases; PI3K: Phosphoinositide 3-kinase; PKC: Protein kinase C.
Tumor suppressor protein p53, one of the most well-known apoptosis inducers, is also one of the sensors of oxidative stress. It also supports cell survival by acting as an antioxidant during cellular stress. p21 protein, another crucial protein participating to cell cycle arrest and apoptosis induction, can affect the transcription of ARE genes. Schmidt et al. showed that cold plasma promotes Nrf2- and p53-mediated granulation and reepithelization in the skin of SKH1 mice. Levels of proinflammatory cytokines IL-6 or TNFα and angiogenetic factors such as FGF, KGF, VEGF and COX2 increase in the early stages of wound healing and this increase depends on Nrf2 signal activation [67]. This observation also indicates that mild inflammation is necessary to trigger wound healing [35].
Recent studies have made significant contributions to elucidate the relationship between the Nrf2/Keap1 signaling pathway and wound healing. However, some points still need to be clarified. In particular, we know very little about the interaction and regulatory mechanisms between signal pathways that trigger autophagy and the Nrf2 signaling. In case of tissue damage and severe ROS production, Nrf2 mediates the prevention of apoptosis of keratinocytes caused by the activation of cytoprotective genes. It is well accepted that Nrf2 acts as a defense signal in the case of oxidative stress and plays a cell protective role by reducing ROS. However, there are studies showing that continuous activation of Nrf2 can also trigger tumorigenesis. Given the dualistic role of Nrf2, many factors such as the contribution of additional signaling pathways, activators and suppressors or even inflammatory mediators should be taken into account to understand how the Nrf2 pathway regulates the wound healing activity. However, formulations to be developed using Nrf2 activators in acute or chronic wounds are seen as a promising treatment.
References
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525.
- Gonzalez, A.C.; Costa, T.F.; Andrade, Z.A.; Medrado, A.R. Wound healing—A literature review. An. Bras. Dermatol. 2016, 91, 614–620.
- Victor, P.; Sarada, D.; Ramkumar, K.M. Pharmacological activation of Nrf2 promotes wound healing. Eur. J. Pharmacol. 2020, 886, 173395.
- Hiebert, P.; Werner, S. Regulation of wound healing by the NRF2 transcription factor—More than cytoprotection. Int. J. Mol. Sci. 2019, 20, 3856.
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes. Dev. 1999, 13, 76–86.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 2020, 25, 5474.
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell Biol. 2003, 23, 8137–8151.
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.-I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045.
- Negi, C.K.; Jena, G. Nrf2, a novel molecular target to reduce type 1 diabetes associated secondary complications: The basic considerations. Eur. J. Pharmacol. 2019, 843, 12–26.
- Kwak, M.-K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145.
- Senger, D.R.; Cao, S. Diabetic wound healing and activation of Nrf2 by herbal medicine. J. Nat. Sci. 2016, 2, e247.
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93.
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4.
- Covas, G.; Marinho, H.S.; Cyrne, L.; Antunes, F. Activation of Nrf2 by H2O2. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; pp. 157–171.
- Li, C.-Q.; Kim, M.Y.; Godoy, L.C.; Thiantanawat, A.; Trudel, L.J.; Wogan, G.N. Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14547–14551.
- Wu, K.; McDonald, P.; Liu, J.; Klaassen, C. Screening of natural compounds as activators of the Keap1-Nrf2 pathway. Planta Med. 2014 80, 97–104.
- Muri, J.; Wolleb, H.; Broz, P.; Carreira, E.M.; Kopf, M. Electrophilic Nrf2 activators and itaconate inhibit inflammation at low dose and promote IL-1β production and inflammatory apoptosis at high dose. Redox. Biol. 2020, 36, 101647.
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and other nutrigenomic nrf2 activators: Can the clinician’s expectation be matched by the reality? Oxid. Med. Cell Longev. 2016, 1–17.
- Lambers Heerspink, H.J.; Fioretto, P.; de Zeeuw, D. Pathogenesis, pathophysiology, and treatment of diabetic nephropathy. In National Kidney Foundation Primer on Kidney Diseases; Elsevier: Amsterdam, The Netherlands, 2014; pp. 222–234.
- Beer, H.D.; Gassmann, M.G.; Munz, B.; Steiling, H.; Engelhardt, F.; Bleuel, K.; Werner, S. Expression and function of keratinocyte growth factor and activin in skin morphogenesis and cutaneous wound repair. J. Investig. Dermatol. Symp. Proc. 2000, 5, 34–39.
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; auf dem Keller, U.; Born-Berclaz, C.; Chan, K.; Kan, Y.W.; Werner, S. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell Biol. 2002, 22, 5492–5505.
- Wang, H.; Pan, L.; Xu, R.; Si, L.; Zhang, X. The molecular mechanism of Nrf2-Keap1 signaling pathway in the antioxidant defense response induced by BaP in the scallop Chlamys farreri. Fish Shellfish Immunol. 2019, 92, 489–499.
- Wruck, C.J.; Götz, M.E.; Herdegen, T.; Varoga, D.; Brandenburg, L.-O.; Pufe, T. Kavalactones protect neural cells against amyloid β peptide-induced neurotoxicity via extracellular signal-regulated kinase 1/2-dependent nuclear factor erythroid 2-related factor 2 activation. Mol. Pharmacol. 2008, 73, 1785–1795.
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99.
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1–NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355.
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71.
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. MCB 2010, 30, 3275–3285.
- Ichimura, Y.; Waguri, S.; Sou, Y.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631.
- Hashimoto, K.; Simmons, A.N.; Kajino-Sakamoto, R.; Tsuji, Y.; Ninomiya-Tsuji, J. TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxid. Redox Signal. 2016, 25, 953–964.
- Ambrozova, N.; Ulrichova, J.; Galandakova, A. Models for the study of skin wound healing. The role of Nrf2 and NF-κB. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub. 2017, 161, 1–13.
- Kubo, Y.; Wruck, C.J.; Fragoulis, A.; Drescher, W.; Pape, H.C.; Lichte, P.; Fischer, H.; Tohidnezhad, M.; Hildebrand, F.; Pufe, T.; et al. Role of Nrf2 in fracture healing: Clinical aspects of oxidative stress. Calcif. Tissue Int. 2019, 105, 341–352.
- Cano Sanchez, M.; Lancel, S.; Boulanger, E.; Neviere, R. Targeting oxidative stress and mitochondrial dysfunction in the treatment of impaired wound healing: A systematic review. Antioxidants 2018, 7, 98.
- Schmidt, A.; Bekeschus, S. Redox for repair: Cold physical plasmas and Nrf2 signaling promoting wound healing. Antioxidants 2018, 7, 146.
- Lippross, S.; Beckmann, R.; Streubesand, N.; Ayub, F.; Tohidnezhad, M.; Campbell, G.; Kan, Y.W.; Horst, F.; Sönmez, T.T.; Varoga, D.; et al. Nrf2 deficiency impairs fracture healing in mice. Calcif. Tissue Int. 2014, 95, 349–361.
- Sun, Y.-X.; Li, L.; Corry, K.A.; Zhang, P.; Yang, Y.; Himes, E.; Mihuti, C.L.; Nelson, C.; Dai, G.; Li, J. Deletion of Nrf2 reduces skeletal mechanical properties and decreases load-driven bone formation. Bone 2015, 74, 1–9.
- Hu, K.; Olsen, B.R. The roles of vascular endothelial growth factor in bone repair and regeneration. Bone 2016, 91, 30–38.
- Kweider, N.; Fragoulis, A.; Rosen, C.; Pecks, U.; Rath, W.; Pufe, T.; Wruck, C.J. Interplay between vascular endothelial growth factor (VEGF) and nuclear factor erythroid 2-related factor-2 (Nrf2): Implications for preeclampsia. J. Biol. Chem. 2011, 286, 42863–42872.
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. Activation of KEAP1/NRF2 stress signaling involved in the molecular basis of hemin-induced cytotoxicity in human pro-erythroid K562 cells. Biochem. Pharmacol. 2020, 175, 113900.
- Panich, U.; Sittithumcharee, G.; Rathviboon, N.; Jirawatnotai, S. Ultraviolet radiation-induced skin aging: The role of DNA damage and oxidative stress in epidermal stem cell damage mediated skin aging. Stem. Cells Int. 2016, 2016, 7370642.
- Schafer, M.; Dutsch, S.; auf dem Keller, U.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes. Dev. 2010, 24, 1045–1058.
- Wu, R.; Zhang, H.; Zhao, M.; Li, J.; Hu, Y.; Fu, J.; Pi, J.; Wang, H.; Xu, Y. Nrf2 in keratinocytes protects against skin fibrosis via regulating epidermal lesion and inflammatory response. Biochem. Pharmacol. 2020, 174, 113846.
- Rabbani, P.S.; Ellison, T.; Waqas, B.; Sultan, D.; Abdou, S.; David, J.A.; Cohen, J.M.; Gomez-Viso, A.; Lam, G.; Kim, C.; et al. Targeted Nrf2 activation therapy with RTA 408 enhances regenerative capacity of diabetic wounds. Diabetes Res. Clin. Pract. 2018, 139, 11–23.
- Hayashi, R.; Himori, N.; Taguchi, K.; Ishikawa, Y.; Uesugi, K.; Ito, M.; Duncan, T.; Tsujikawa, M.; Nakazawa, T.; Yamamoto, M.; et al. The role of the Nrf2-mediated defense system in corneal epithelial wound healing. Free Radic. Biol. Med. 2013, 61, 333–342.
- Ayuk, S.M.; Abrahamse, H.; Houreld, N.N. The Role of Matrix Metalloproteinases in Diabetic Wound Healing in relation to Photobiomodulation. J Diabetes Res. 2016, 2897656.
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967.
- Zampetaki, A.; Kirton, J.P.; Xu, Q. Vascular repair by endothelial progenitor cells. Cardiovasc. Res. 2008, 78, 413–421.
- António, N.; Fernandes, R.; Soares, A.; Soares, F.; Lopes, A.; Carvalheiro, T.; Paiva, A.; Pêgo, G.M.; Providência, L.A.; Gonçalves, L.; et al. Reduced levels of circulating endothelial progenitor cells in acute myocardial infarction patients with diabetes or pre-diabetes: Accompanying the glycemic continuum. Cardiovasc. Diabetol. 2014, 13, 1012014.
- Tsukada, S.; Masuda, H.; Jung, S.Y.; Yun, J.; Kang, S.; Kim, D.Y.; Park, J.H.; Ji, S.T.; Kwon, S.M.; Asahara, T. Impaired development and dysfunction of endothelial progenitor cells in type 2 diabetic mice. Diabetes Metab. 2017, 43, 154–162.
- Fadini, G.P.; Sartore, S.; Schiavon, M.; Albiero, M.; Baesso, I.; Cabrelle, A.; Agostini, C.; Avogaro, A. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia 2006, 49, 3075–3084.
- Ingram, D.A.; Lien, I.Z.; Mead, L.E.; Estes, M.; Prater, D.N.; Derr-Yellin, E.; DiMeglio, L.A.; Haneline, L.S. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes 2008, 57, 724–731.
- Wang, R.Y.; Liu, L.H.; Liu, H.; Wu, K.F.; An, J.; Wang, Q.; Liu, Y.; Bai, L.J.; Qi, B.M.; Qi, B.L.; et al. Nrf2 protects against diabetic dysfunction of endothelial progenitor cells via regulating cell senescence. Int. J. Mol. Med. 2018, 42, 1327–1340.
- Florczyk, U.; Jazwa, A.; Maleszewska, M.; Mendel, M.; Szade, K.; Kozakowska, M.; Grochot-Przeczek, A.; Viscardi, M.; Czauderna, S.; Bukowska-Strakova, K.; et al. Nrf2 regulates angiogenesis: Effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid. Redox Signal. 2014, 20, 1693–1708.
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. A New Human Blood-Retinal Barrier Model Based on Endothelial Cells, Pericytes, and Astrocytes. Int. J. Mol. Sci. 2020, 21, 1636.
- Villarreal-Ponce, A.; Tiruneh, M.W.; Lee, J.; Guerrero-Juarez, C.F.; Kuhn, J.; David, J.A.; Dammeyer, K.; Mc Kell, R.; Kwong, J.; Rabbani, P.S.; et al. Keratinocyte-Macrophage Crosstalk by the Nrf2/Ccl2/EGF Signaling Axis Orchestrates Tissue Repair. Cell Rep. 2020, 33, 108417.
- Long, M.; Rojo de la Vega, M.; Wen, Q.; Bharara, M.; Jiang, T.; Zhang, R.; Zhou, S.; Wong, P.K.; Wondrak, G.T.; Zheng, H.; et al. An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes 2016, 65, 780–793.
- Valcarcel-Ares, M.N.; Gautam, T.; Warrington, J.P.; Bailey-Downs, L.; Sosnowska, D.; de Cabo, R.; Losonczy, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: Implications for microvascular aging. J. Gerontol. Series A Biol. Sci. Med. Sci. 2012, 67, 821–829.
- Kuhn, J.; Sultan, D.L.; Waqas, B.; Ellison, T.; Kwong, J.; Kim, C.; Hassan, A.; Rabbani, P.S.; Ceradini, D.J. Nrf2-activating Therapy Accelerates Wound Healing in a Model of Cutaneous Chronic Venous Insufficiency. Plast. Reconstr. Surg. 2020, 8, e3006.
- Chen, B.; Sun, Y.; Zhang, J.; Zhu, Q.; Yang, Y.; Niu, X.; Deng, Z.; Li, Q.; Wang, Y. Human embryonic stem cell-derived exosomes promote pressure ulcer healing in aged mice by rejuvenating senescent endothelial cells. Stem. Cell Res. Ther. 2019, 10, 142.
- Chen, N.; Li, Y.; Huang, N.; Yao, J.; Luo, W.F.; Jiang, Q. The Nrf2 activator MIND4-17 protects retinal ganglion cells from high glucose-induced oxidative injury. J. Cell Physiol. 2020, 235, 7204–7213.
- Wu, J.; Sun, X.; Jiang, Z.; Jiang, J.; Xu, L.; Tian, A.; Sun, X.; Meng, H.; Li, Y.; Huang, W.; et al. Protective role of NRF2 in Macrovascular Complications of Diabetes. J. Cell Mol. Med. 2020, 16, 8903–8917.
- Li, S.; Yang, H.; Chen, X. Protective effects of sulforaphane on diabetic retinopathy: Activation of the Nrf2 pathway and inhibition of NLRP3 inflammasome formation. Exp. Anim. 2019, 68, 221–231.
- Li, D.; Wu, C.; Mao, L.; Gao, Z.; Xia, N.; Liu, C.; Mei, X. LPS-stimulated Macrophage Exosomes Inhibit Inflammation by Activating the Nrf2/HO-1 Defense Pathway and Promote Wound Healing in Diabetic Rats. Res. Sq. 2020.
- Fan, J.; Liu, H.; Wang, J.; Zeng, J.; Tan, Y.; Wang, Y.; Yu, X.; Li, W.; Wang, P.; Yang, Z.; et al. Procyanidin B2 improves endothelial progenitor cell function and promotes wound healing in diabetic mice via activating Nrf2. J. Cell Mol. Med. 2021, 25, 652–665.
- Soares, M.A.; Cohen, O.D.; Low, Y.C.; Sartor, R.A.; Ellison, T.; Anil, U.; Anzai, L.; Chang, J.B.; Saadeh, P.B.; Rabbani, P.S.; et al. Restoration of Nrf2 Signaling Normalizes the Regenerative Niche. Diabetes 2016, 65, 633–646.
- Schmidt, A.; von Woedtke, T.; Vollmar, B.; Hasse, S.; Bekeschus, S. Nrf2 signaling and inflammation are key events in physical plasma-spurred wound healing. Theranostics 2019, 9, 1066–1084.

