 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Annalisa Marcuzzi | + 1908 word(s) | 1908 | 2021-05-08 10:11:52 | | | |
| 2 | Vivi Li | Meta information modification | 1908 | 2021-05-11 03:26:40 | | |
Video Upload Options
Mitoquinone (MitoQ) is a mitochondrial reactive oxygen species scavenger that is characterized by high bioavailability. Prior studies have demonstrated its neuroprotective potential. Indeed, the release of reactive oxygen species due to damage to mitochondrial components plays a pivotal role in the pathogenesis of several neurodegenerative diseases.
1. Introduction
The mitochondria represent the main sources of reactive oxygen species (ROS) related to several diseases caused by alteration of cholesterol biosynthesis [1][2][3]. In the past years, new drugs belonging to the Mitochondria-Targeted Antioxidants (MTAs) family, such as Mitoquinone (MitoQ), Mitotempo and MitoVitE, have proven effective in decreasing oxidative stress across multiple disease models. MTAs are indeed known for their activity against the activation of the mitochondrial damage because of their antioxidant and mitochondrial protective properties. In particular, MitoQ is characterized by high bioavailability, and several prior studies have demonstrated its neuroprotective potential [4][5].
Of note, although the FDA (U.S. Food and Drug Administration) has not yet approved MitoQ as a drug, 19 studies are currently registered to evaluate the effect of MitoQ in several diseases, such as multiple sclerosis, Parkinson’s disease, Alzheimer’s disease and cystic fibrosis [6].
Cholesterol in cells is essential for the correct organization of the cell membrane, determines its permeability and fluidity and is considered an important factor that regulates the plasticity of membranes [7]. This is especially important for mitochondria, which are considered the energy source of the cell, in which cholesterol is fundamental to membrane maintenance [8][9][10]. Moreover, the cholesterol homeostasis in the mitochondria is necessary to ensure and maintain the mitochondrial biogenesis [11]. Several works have provided evidence about the regulation of microtubule-based mitochondrial trafficking, highlighting that mitochondrial motility affects neuronal growth and synapses [12][13]. Consequently, the mitochondrial dynamics are particularly sensitive to various stimuli and the mechanism underlying its regulation is a balance between mitochondrial fusion and fission [14]. The alterations or perturbations in mitochondrial dynamics can been observed in several neurodegenerative diseases [15][16].
These dysregulations are more easily detectable in the central nervous system, since this area represents a system closed to cholesterol synthesis [17]. It is well known that statins, such as lovastatin, are able to induce apoptosis in neuronal cell lines and that the mevalonate prevents the programmed cell death induced by the blockade of this pathway [18][19].
In previous studies, indeed, we demonstrated that in neuronal cells the blockade of the cholesterol pathway, also called mevalonate pathway, using a statin, induced significant morphological changes and dysfunctions in mitochondria [20] and that the presence of antioxidant MitoQ plays a fundamental role as preventive agent [17].
To date, the molecular mechanism undergoing the MitoQ role in this experimental cellular model is not known and this is an important step to improve our knowledge for several pathogenesis linked to cholesterol metabolism (Figure 1).
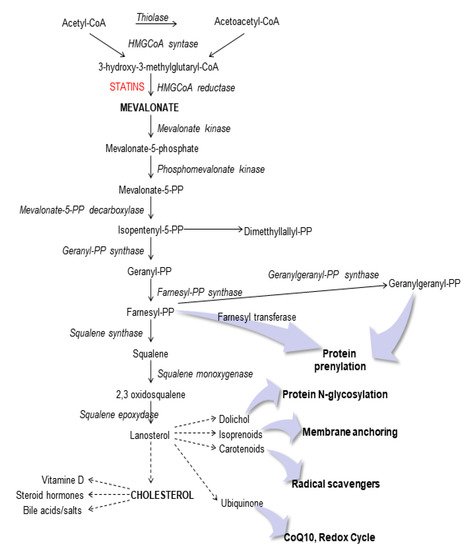
Figure 1. Schematic representation of the mevalonate pathway.
It has been shown that blocking of the pathway of cholesterol metabolism is associated with increased susceptibility to pro-inflammatory stimuli which induces the activation of inflammation.
Important regulators of the inflammatory response are represented by the inflammasomes, cytosolic multiprotein complexes that mediates the activation of the pro-inflammatory caspase 1 [21][22]. These enzymes are key regulators of the conversion of the inactive pro-IL-1β into the mature active IL-1β and promote pyroptosis, a form of programmed cell death. The most investigated inflammasome is NLRP3 (NACHT, LRR and PYD domains-containing protein 3), which has been associated with various diseases considering that caspase 1 activation is crucial for starting inflammation in response to injury or infection [23]. In general, one of the most important regulator of inflammasomes is represented by autophagy, a complex mechanism that maintains cellular homeostasis by the degradation of unnecessary or dysfunctional components through the action of lysosomes. Several studies have established a strict interplay between the inflammasome and autophagy, and the balance between these systems is crucial for various biological functions, such as inflammation, metabolism, programmed cell death and cell differentiation [24][25][26].
2. Effect of MitoQ on Mitochondrial Electrochemical Potential Gradient
To set the experimental conditions and verify the mitochondrial damages triggered from the blockade of metabolic pathway, we employed a cytofluorimetric assay, using the lipophilic membrane-permeant JC-1 dye that accumulates in mitochondria and detects mitochondrial depolarization occurring in a variety of conditions, including apoptosis [27][28]. JC-1 staining confirmed that the statin caused significant damage to mitochondrial membrane after an extended incubation (48 h, Lova: 73.45 ± 14.61 vs. untreated: 24.96 ± 9.41; *** p < 0.0001), not so evident in the short period (12 h, Lova: 27.87 ± 0.81 vs. untreated: 17.17 ± 2.25; * p < 0.05) (24 h, Lova: 35.38 ± 3.52 vs. untreated: 23.20 ± 4.28; * p < 0.05). The presence of MitoQ limited apoptosis of DAOY cells in the early-stage analysis (12 h, MitoQ + Lova: 14.08 ± 1.05 vs. Lova: 28.30 ± 0.42; §§ p < 0.01) and not at 24 and 48 h (Figure 2). Moreover, the data show that MitoQ alone is comparable to untreated condition.
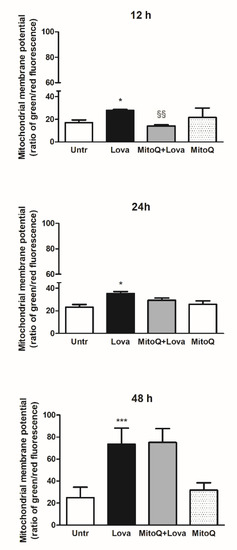
Figure 2. JC-1, a lipophilic cation dye, staining to evidence the mitochondrial membrane potential (N = 4 independent experiments). Data are shown as means ± SD (Lova vs. Untreated: * p < 0.05; *** p < 0.001; MitoQ + Lova vs. Lova: §§ p < 0.01 based on one-way ANOVA, post-test Bonferroni). Abbreviations: Untr, untreated; Lova, lovastatin.
3. Inflammatory Genes Expression Related to MitoQ Activity
To investigate the activation of inflammatory platforms linked to apoptotic systems, we measured the expression levels of NLRP3, CASP-1 (caspase 1) and OPA-1 genes by qPCR in our experimental cellular model. Figure 3 shows that lovastatin treatment for 12 and 24 h induced a hyper-expression of NLRP3, CASP-1 and OPA-1 compared to the untreated condition. Although less evident, the same trend is detectable after 48 h of lovastatin challenge only for OPA-1 gene, but not for NLRP3 and CASP-1 genes.
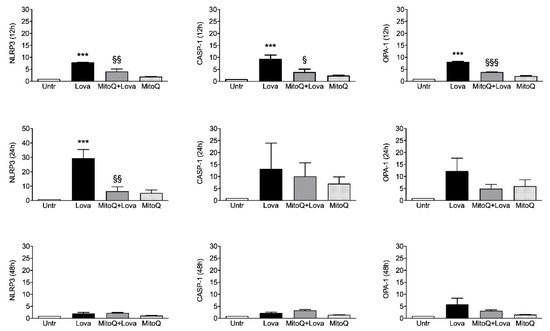
Figure 3. Relative quantification by qPCR of NLRP3, OPA-1 and CASP-1 genes in DAOY cell line (N = 4 independent experiments). DAOY cells were incubated for 12, 24 and 48 h with lovastatin and Mitoq and afterward collected for RNA extraction and quantification. Results are reported as mean ± SD. Statistically significant p-values are shown (Lova vs. Untreated: *** p < 0.001; MitoQ + Lova vs. Lova: § p < 0.05; §§ p < 0.01; §§§ p < 0.001 based on one-way ANOVA, post-test Bonferroni). Abbreviations: Untr, untreated; Lova, lovastatin; NLRP3, NACHT, LRR and PYD domains-containing protein 3; OPA-1, Optic Atrophy 1; CASP-1, caspase 1.
The 12 h combined treatment of lovastatin and MitoQ reduced NLRP3, CASP-1 and OPA-1 gene expression levels in comparison with lovastatin alone; the downward trend was maintained for NLRP3 even if it was less evident for CASP-1 and OPA-1 at the 24 h challenge. These results are not confirmed after 48 h of the combined treatment: NLRP3 and CASP-1 expression levels can be compared with the condition of lovastatin alone, while the decreasing trend of OPA-1 expression is slightly appreciable.
Taken together, these results confirm that the addition of MitoQ to lovastatin is able to limit the inflammation platform in our cellular model in a short treatment timeframe (12 and 24 h). Moreover, we can assume that 48 h is a too long stimulation timepoint for our experimental design.
4. Discussion
The cholesterol in the brain represents approximately 25% of the total cholesterol present in our body, containing about 10-fold more than any organ [29]. In the central nervous system, high cholesterol levels are essential in the myelin sheath to facilitate the transmission of electric signals and it is also necessary for synapse and dendrite formation. Furthermore, this sterol is fundamental for neuronal physiology, both during development and in the adult stage. Notably, several disorders are associated with defects in cholesterol synthesis and metabolism; there are, indeed, a large family of diseases caused by inborn errors of metabolism and including rare pathologies such as Niemann–Pick C disease, mevalonate kinase deficiency and Smith–Lemli–Optiz syndrome [30][31]. In particular, most of the pathogenesis of these diseases suggest that there is a common involvement of the inflammation platform [32]. The NLRP3 inflammasome represents a physiological trigger to activate the inflammation, as confirmed by the results obtained after 12 and 24 h of treatment with lovastatin. It is therefore not surprising that in this study caspase 1 (CASP-1) showed a comparable activation trend at 12 h, and that the addition of MitoQ was able to modulate and decrease this expression due to the blockade of the pathway. These data were statistically significant 12 h after the stimulus: MitoQ, indeed, was able to prevent or limit the inflammation activity, but this ability decreased at later timepoints (24 and 48 h).
The total absence of effects in long-time treatments could be explained by the MitoQ half-life or by the severe cellular morphological changes and damage due to lovastatin, a condition in which MitoQ action is not effective. The homogeneous cytokines profile at 12 h confirms that at this timepoint the MitoQ was able to counteract the inflammatory activation supported by gene expression. Data obtained with the membrane-permeant JC-1 dye, used to monitor mitochondrial health, confirm that the mitochondrial membrane was damaged after 48 h of lovastatin, regardless of MitoQ treatment [33]. In contrast, at 12 h, we can observe that MitoQ is able to counteract the lovastatin treatment, supporting the hypothesis that MitoQ can modulate the morphologic change in a short time.
It is important, instead, to highlight that the high significant OPA-1 expression level after 12 h of lovastatin treatment is strictly associated with its biological role in the regulation of mitochondrial stability and cellular energy output, as demonstrated in previous studies [34][35]. Indeed, OPA-1 is a mitochondrial fusion protein which is characterized by rigorous cellular regulation, including apoptosis and respiratory capacity, necessary for oxidative phosphorylation (OXPHOS) and ATP production [36][37][38].
The obtained data are notably relevant in all time analyzed since lovastatin treatment in OXPHOS-Complex V induced a significant increase in comparison with untreated condition. It should be noted that MitoQ at 48 h was able to reduce the Complex V activity: these results can be interpreted as a physiological mitochondrial mechanism to protect the membranes. Notoriously, OXPHOS-Complex V, also called ATP synthase, is the final enzyme in the oxidative phosphorylation pathway and represents the energy source of the system. This evidence suggests that the ATP synthase is correlated with the fusion of the mitochondrial inner membrane regulated by OPA-1 [35]. The fundamental role of this protein in mitochondria stability and in the energy production process is attested by its deficiency in the most common age-related neurodegenerative diseases such as Alzheimer’s diseases and Parkinson’s disease [34][35][39][40]
Moreover, literature data demonstrate that OXPHOS activity was correlated to NLRP3 inflammasome activation in several diseases [41][42][43][44] so much that NLRP3 may be considered as a novel biomarker and therapeutic target for these pathologies [45].
It is interesting to highlight that just NLRP3 expression remains at 24 h, and these data were confirmed by IL-1α secretion, validating the timepoints that interplay between these phenomena and supporting the role of inflammasome in neurodegenerative diseases [23][46]. The same trend is found with IL-6 and TNF-α, which together with IL-1β are considered as key players of the neuroinflammation, since they are the interface between the immune system and the nervous system. These cytokines, indeed, are released in response to multiple stimuli able to activate the immune response and interact with the activity of glial and neuronal cells, modulating for example the sensitivity of synapses to the activation of membrane receptors for glutamate, GABA or endocannabinoids and regulating the release of neurotransmitters such as dopamine [47][48]. Moreover, it is interesting to highlight the modulation of IFN-γ, known to promote the expression of TNF-α and IL-1 β in microglia, which suggests a key role of this cytokine in the development of immune and inflammatory responses involved in neurodegeneration mechanisms in the CNS [49][50][51]. Moreover, previous studies have revealed IL-17 as a cytokine involved in chronic inflammatory neurological diseases, and we can confirm that, in our neuronal model, it can be modulated from the inflammation [52][53]. All these data are relevant for the proper understanding of cytokines signaling pathways involved in the regulation of neuroinflammation and could be crucial for the development of new therapeutical strategies [54].
References
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214.
- Martín, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO Rep. 2014, 15, 1036–1052.
- Johannsen, D.L.; Ravussin, E. The role of mitochondria in health and disease. Curr. Opin. Pharmacol. 2009, 9, 780–786.
- Williamson, J.; Davison, G. Targeted Antioxidants in Exercise-Induced Mitochondrial Oxidative Stress: Emphasis on DNA Damage. Antioxidants 2020, 9, 1142.
- Battogtokh, G.; Choi, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Huh, K.M.; Cho, Y.-Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880.
- Clinical Trials. Available online: (accessed on 25 April 2021).
- Gliozzi, M.; Musolino, V.; Bosco, F.; Scicchitano, M.; Scarano, F.; Nucera, S.; Zito, M.C.; Ruga, S.; Carresi, C.; Macrì, R.; et al. Cholesterol homeostasis: Researching a dialogue between the brain and peripheral tissues. Pharmacol. Res. 2021, 163, 105215.
- Martin, L.A.; Kennedy, B.E.; Karten, B. Mitochondrial cholesterol: Mechanisms of import and effects on mitochondrial function. J. Bioenerg. Biomembr. 2014, 48, 137–151.
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93.
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666.
- Schwarz, T.L. Mitochondrial Trafficking in Neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304.
- Sheng, Z.-H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098.
- Rangaraju, V.; Lewis, T.L., Jr.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.-K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208.
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681.
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22.
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; De La Cruz-Ojeda, P.; De La Mata, M.; Cotán, D.; Oropesa-Ávila, M.; De Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1.
- Marcuzzi, A.; Loganes, C.; Valencic, E.; Piscianz, E.; Monasta, L.; Bilel, S.; Bortul, R.; Celeghini, C.; Zweyer, M.; Tommasini, A. Neuronal Dysfunction Associated with Cholesterol Deregulation. Int. J. Mol. Sci. 2018, 19, 1523.
- Macaulay, R.J.; Wang, W.; Dimitroulakos, J.; Becker, L.E.; Yeger, H. Lovastatin-induced apoptosis of human medulloblastoma cell lines in vitro. J. Neurooncol. 1999, 42, 1–11.
- Wang, W.; Macaulay, R.J. Mevalonate Prevents Lovastatin-Induced Apoptosis in Medulloblastoma Cell Lines. Can. J. Neurol. Sci. 1999, 26, 305–310.
- Marcuzzi, A.; Tricarico, P.M.; Piscianz, E.; Kleiner, G.; Brumatti, L.V.; Crovella, S. Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis. 2013, 4, e585.
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15.
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247.
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953.
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803.
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714.
- Yuk, J.-M.; Jo, E.-K. Crosstalk between Autophagy and Inflammasomes. Mol. Cells 2013, 36, 393–399.
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol 2019, 9, e3128.
- Perelman, A.; Wachtel, C.; Cohen, M.E.; Haupt, S.; Shapiro, H.M.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430.
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264.
- Björkhem, I.; Leoni, V.; Meaney, S. Genetic connections between neurological disorders and cholesterol metabolism. J. Lipid Res. 2010, 51, 2489–2503.
- Marzban, H.; Kong, J.; Mehr, S.; Vriend, J.; Li, J.; Guan, T.; Chung, S.; Mirzaei, N.; Marzban, A.; Shojaei, S.; et al. Mevalonate Cascade and Neurodevelopmental and Neurodegenerative Diseases: Future Targets for Therapeutic Application. Curr. Mol. Pharmacol. 2017, 10, 115–140.
- Platt, F.M.; Wassif, C.; Colaco, A.; Dardis, A.; Lloyd-Evans, E.; Bembi, B.; Porter, F.D. Disorders of cholesterol metabolism and their unanticipated convergent isms omechanf disease. Annu. Rev. Genom. Hum. Genet. 2014, 15, 173–194.
- Colina-Tenorio, L.; Horten, P.; Pfanner, N.; Rampelt, H. Shaping the mitochondrial inner membrane in health and disease. J. Intern. Med. 2020, 287, 645–664.
- Park, J.-B.; Nagar, H.; Choi, S.; Jung, S.-B.; Kim, H.-W.; Kang, S.K.; Lee, J.W.; Lee, J.H.; Park, J.-W.; Irani, K.; et al. IDH2 deficiency impairs mitochondrial function in endothelial cells and endothelium-dependent vasomotor function. Free Radic. Biol. Med. 2016, 94, 36–46.
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101.
- Xie, T.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Wu, J.; Sun, H. CoenzymeQ10-Induced Activation of AMPK-YAP-OPA1 Pathway Alleviates Atherosclerosis by Improving Mitochondrial Function, Inhibiting Oxidative Stress and Promoting Energy Metabolism. Front. Pharmacol. 2020, 11, 1034.
- Garcia, I.; Innis-Whitehouse, W.; Lopez, A.; Keniry, M.; Gilkerson, R. Oxidative insults disrupt OPA1-mediated mitochondrial dynamics in cultured mammalian cells. Redox Rep. 2018, 23, 160–167.
- Landes, T.; Leroy, I.; Bertholet, A.; Diot, A.; Khosrobakhsh, F.; Daloyau, M.; Davezac, N.; Miquel, M.-C.; Courilleau, D.; Guillou, E.; et al. OPA1 (dys)functions. Semin. Cell Dev. Biol. 2010, 21, 593–598.
- Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules 2020, 10, 1437.
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216.
- Reinecke, F.; Smeitink, J.A.; van der Westhuizen, F.H. XPHOS gene expression and control in mitochondrial disorders. Biochim Biophys. Acta 2009, 1792, 1113–1121.
- Tang, J.X.; Thompson, K.; Taylor, R.W.; Oláhová, M. Mitochondrial OXPHOS Biogenesis: Co-Regulation of Protein Synthesis, Import, and Assembly Pathways. Int. J. Mol. Sci. 2020, 21, 3820.
- Chung, I.-C.; Chen, L.-C.; Tsang, N.-M.; Chuang, W.-Y.; Liao, T.-C.; Yuan, S.-N.; OuYang, C.-N.; Ojcius, D.M.; Wu, C.-C.; Chang, Y.-S. Mitochondrial Oxidative Phosphorylation Complex Regulates NLRP3 Inflammasome Activation and Predicts Patient Survival in Nasopharyngeal Carcinoma. Mol. Cell. Proteom. 2020, 19, 142–154.
- He, Z.-F.; Jin, X.-R.; Lin, J.-J.; Zhang, X.; Liu, Y.; Xu, H.-L.; Xie, D.-Y. NALP3 orchestrates cellular bioenergetics to facilitate non-small cell lung cancer cell growth. Life Sci. 2020, 241, 117165.
- Bernardi, S.; Marcuzzi, A.; Piscianz, E.; Tommasini, A.; Fabris, B. The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 4058.
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248.
- Cacquevel, M.; Lebeurrier, N.; Chéenne, S.; Vivien, D. Cytokines in neuroinflammation and Alzheimer’s disease. Curr. Drug Targets 2004, 5, 529–534.
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82.
- Bartfai, T.; Schultzberg, M. Cytokines in neuronal cell types. Neurochem. Int. 1993, 22, 435–444.
- Benveniste, E.N. Cytokine circuits in brain. Implications for AIDS dementia complex. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1994, 72, 71–88.
- Porrini, A.M.; Reder, A.T. IFN-gamma, IFN-beta, and PGE1 affect monokine secretion: Relevance to monocyte activation in multiple sclerosis. Cell Immunol. 1994, 157, 428–438.
- Milovanovic, J.; Arsenijevic, A.; Stojanovic, B.; Kanjevac, T.; Arsenijevic, D.; Radosavljevic, G.; Milovanovic, M.; Arsenijevic, N. Interleukin-17 in Chronic Inflammatory Neurological Diseases. Front. Immunol. 2020, 11, 947.
- Sun, C.; Zhang, J.; Chen, L.; Liu, T.; Xu, G.; Li, C.; Yuan, W.; Xu, H.; Su, Z. IL-17 contributed to the neuropathic pain following peripheral nerve injury by promoting astrocyte proliferation and secretion of proinflammatory cytokines. Mol. Med. Rep. 2016, 15, 89–96.
- Chen, R.; Yin, C.; Fang, J.; Liu, B. The NLRP3 inflammasome: An emerging therapeutic target for chronic pain. J. Neuroinflammation 2021, 18, 1–12.

