 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marc Herb | + 9010 word(s) | 9010 | 2021-04-23 11:22:22 | | | |
| 2 | Catherine Yang | -86 word(s) | 8924 | 2021-05-10 09:36:17 | | |
Video Upload Options
Reactive oxygen species (ROS) are a chemically defined group of reactive molecules derived from molecular oxygen. ROS are involved in a plethora of processes in cells in all domains of life, ranging from bacteria, plants and animals, including humans. The importance of ROS for macrophage-mediated immunity is unquestioned. Their functions comprise direct antimicrobial activity against bacteria and parasites as well as redox-regulation of immune signaling and induction of inflammasome activation.
1. Reactive Oxygen Species
The term reactive oxygen species (ROS) describes a group of molecules with at least one oxygen atom and with higher reactivity than molecular oxygen (O2). This group consists of two subclasses: (I) highly reactive free radicals, including the superoxide anion (O2●−), the hydroxyl radical (●OH), alkoxyl (●OOR) and peroxyl radicals (●OOH) [1][2][3] and (II) nonradical species such as hydrogen peroxide (H2O2), singlet oxygen (1O2) [4][5], ozone (O3), and the hypochlorite anion (OCl−) [6][7]. ROS are produced by nearly all organisms and cells [8][9][10][11][12]. O2●−, the common precursor of all ROS produced by cells, is produced by single electron transfer to O2. O2●− quickly dismutates to H2O2, either spontaneously in the presence of water or catalyzed by superoxide dismutases [13][14] (Figure 1).
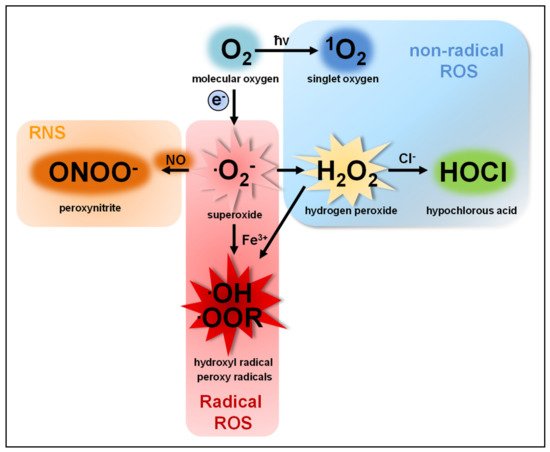
O2− and H2O2 are the two most abundant ROS subspecies in cells, but highly differ in their chemical parameters and, therefore, in their behavior and function.
O2− shows higher reactivity than H2O2 and cannot cross cell membranes, except through ion channels, such as voltage-dependent anion channels (VDAC) [15]. An increase in O2●− levels is associated with oxidative stress and cellular damage [16][17][18][19], such as oxidation of proteins [20][21][22], amino acids [23] and DNA [19][24] or lipid peroxidation [25][26][27]. Moreover, O2●− can irreversibly inactivate proteins and thereby contribute to cellular signaling [28].
H2O2, as a diffusible and relatively stable molecule, is more suitable as a cellular signaling factor than O2●− [29][30][31][32][33]. Although H2O2 is more diffusible than O2●−, its diffusion over membranes is limited [34][35]. This questions the view of saturation of the cell with H2O2 to induce signaling pathways without regarding the compartment of ROS production [36][37]. Aquaporins allow a faster and controlled passage of H2O2 over membranes and add another regulatory level to ROS-mediated signaling [38][39]. The majority of H2O2-mediated signaling is based on the oxidation of cysteine residues [30][40][41][42][43][44][45]. At physiological pH, thiol groups of cysteines (Cys-SH) exposed to the cytosol are deprotonated to thiolate groups (Cys-S−), which are susceptible to oxidation in dependency of their pKa [46][47]. H2O2-dependent signaling occurs at nanomolar concentrations (≈100 nM), leading to reversible oxidation of the thiolate group to a sulfenic group (Cys-SOH). Protein oxidation by H2O2 can lead to allosteric changes that alter binding affinity for substrates or promote or inhibit enzymatic function [48][49][50][51]. Moreover, it can lead to covalent linkage of cysteine residues by disulfide bonds (Cys-S-S-Cys) [30][40][52]. Such protein oxidations can be reversed by the antioxidant defense system and therefore function as important redox switches in various cellular processes [53][54]. Excessive H2O2 production, however, leads to further oxidation of the sulfenic group (Cys-SOH) to sulfinic (Cys-SO2H) and sulfonic groups (Cys-SO3H), a process that is irreversible and results in protein malfunction [55].
2. Macrophages and ROS
Macrophages often are the first immune cells that encounter invading pathogens [56][57][58]. They engulf pathogens, dead cells and cellular debris by phagocytosis and subsequently degrade the cargo in phagolysosomes [59][60][61]. Beyond the production of ROS, macrophages also employ an array of directly antimicrobial mechanisms, e.g., the generation of reactive nitrogen species (RNS) in the phagosome and the delivery of cathepsins and other hydrolases into maturing phagosomes [62][63][64][65][66][67][68]. Indirect antimicrobial mechanisms include the activation of inflammasomes and the secretion of cytokines and chemokines, which help to orchestrate the subsequent innate and adaptive immune responses [30][69], and the MHC-dependent presentation of pathogen-derived antigens [70]. In this review, we will focus on the different ways of how macrophages use ROS for antimicrobial defense.
2.1. Direct Antimicrobial Functions of ROS in Macrophages
2.1.1. ROS vs. Bacteria
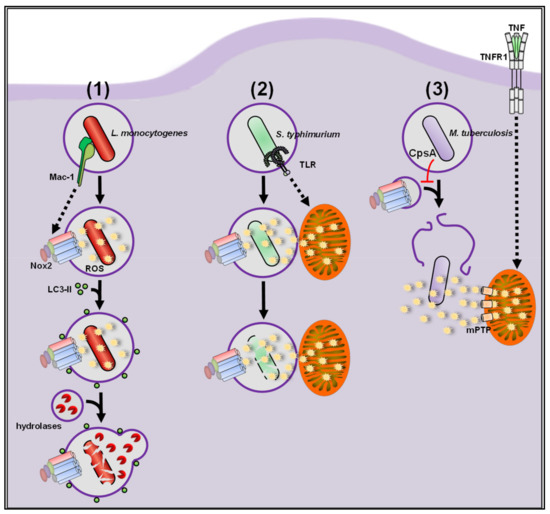
2.1.2. ROS vs. Parasites
2.1.3. ROS vs. Viruses
2.2. Immune-Regulatory Functions of ROS
2.2.1. Immune-Regulatory Functions of mtROS
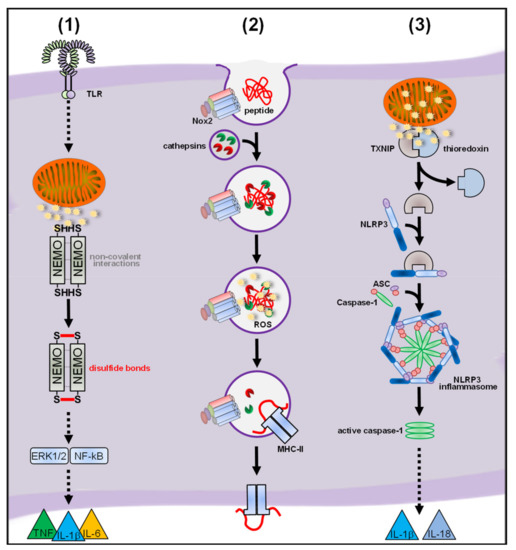
2.2.2. Regulatory Functions of Nox-Derived ROS
2.3. ROS and Inflammasomes
Inflammasomes are cytosolic high-molecular weight complexes that are activated by various different stimuli such as infection, PAMPs and DAMPs. In most cases, inflammasome activation results in assembly of the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), recruitment and autocatalytic activation of caspase-1 and finally release of IL-1β and IL-18. Initiation of pyroptosis, a regulated lytic cell death pathway, can be a consequence of inflammasome activation but is not obligatory. Five receptor proteins leading to inflammasome assembly were identified. The canonical inflammasomes are activated by the nucleotide-binding oligomerization domain (NOD) of the leucine-rich repeat (LRR)-containing protein (NLR) family members NLRP1, NLRP3, NLRC4, NLRP6, NLRP7, NLRP9b, the absent in melanoma 2 (AIM2)-like receptor (ALR) AIM2 and Pyrin [160][161][162][163]. The noncanonical inflammasome, which activates caspase-11 in mice and caspase-4 and/or caspase-5 in humans, is activated through direct recognition of LPS in the cytosol [160]. Other inflammasomes, such as NLRP2, NLRC5, NLRP12, retinoic acid-inducible gene I (RIG-I), and IFNγ-inducible protein 16 (IFI16) also activate caspase-1 but the underlying mechanisms are not yet fully understood [161][164][165][166][167][168]. The crucial role for inflammasome activation during infection of macrophages is well established and has been excellently reviewed elsewhere [161][169][170]. It is also clear that ROS are necessary for inflammasome activation. The underlying molecular mechanisms of ROS-mediated inflammasome activation, however, remain poorly defined. Here, we will focus on studies that investigated the role of ROS during inflammasome activation in macrophages.
2.3.1. Nox-Derived ROS in Inflammasome Activation
2.3.2. mtROS in Inflammasome Activation
3. ROS Probes
3.1. Diffusable ROS Probes (Total Cellular ROS Detection)
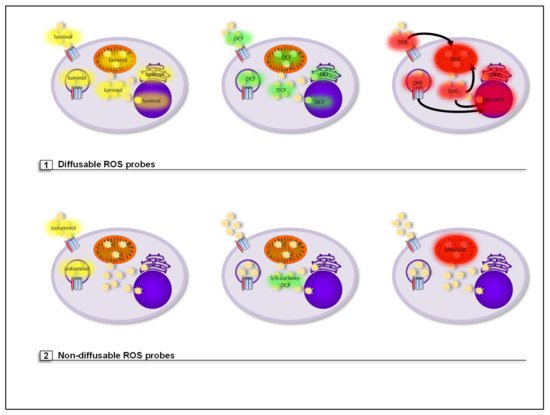
3.2. Nondiffusable ROS Probes (Compartment-Specific ROS Detection)
4. ROS Scavengers
5. ROS Source Inhibitors
6. Concluding Remarks
-
usage of only one type of ROS probe without explanation of the rationale behind the choice, such as compartment or ROS subspecies specificity or no specificity at all (“total cellular ROS”);
-
usage of unspecific inhibitors of ROS production or usage of only globally working ROS scavengers;
-
no genetic evidence for the ROS source (especially in case of Nox enzymes);
-
inconsistent use of different stimuli and macrophage cell types for experiments in one study.
References
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910.
- Haber, F.; Weiss, J.; Pope, W.J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1997, 147, 332–351.
- Prousek, J. Fenton chemistry in biology and medicine. Pure Appl. Chem. 2007, 79, 2325–2338.
- Das, K.; Roychoudhury, A. Reactive oxygen species (ros) and response of antioxidants as ros-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53.
- Hatz, S.; Lambert, J.D.; Ogilby, P.R. Measuring the lifetime of singlet oxygen in a single cell: Addressing the issue of cell viability. Photochem. Photobiol. Sci. 2007, 6, 1106–1116.
- Aratani, Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018, 640, 47–52.
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-mediated regulation of innate and adaptive immunity: The role of myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817.
- Chavez, V.; Mohri-Shiomi, A.; Garsin, D.A. Ce-duox1/bli-3 generates reactive oxygen species as a protective innate immune mechanism in caenorhabditis elegans. Infect. Immun. 2009, 77, 4983–4989.
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999.
- Pendyala, S.; Gorshkova, I.A.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of nox4 and nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid. Redox Signal. 2009, 11, 747–764.
- Ha, E.M.; Oh, C.T.; Ryu, J.H.; Bae, Y.S.; Kang, S.W.; Jang, I.H.; Brey, P.T.; Lee, W.J. An antioxidant system required for host protection against gut infection in drosophila. Dev. Cell 2005, 8, 125–132.
- Levine, A.; Tenhaken, R.; Dixon, R.; Lamb, C. H2o2 from the oxidative burst orchestrates the plant hypersensitive disease resistance response. Cell 1994, 79, 583–593.
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145.
- Cheeseman, K.H.; Slater, T.F. An introduction to free radical biochemistry. Br. Med. Bull. 1993, 49, 481–493.
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. Vdac, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285.
- Gutteridge, J.M.C.; Halliwell, B. Mini-review: Oxidative stress, redox stress or redox success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186.
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell Longev. 2017, 2017, 8416763.
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 2012, 1–26.
- Davies, K.J. Adaptive homeostasis. Mol. Asp. Med. 2016, 49, 1–7.
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316.
- Griffiths, H.R.; Dias, I.H.; Willetts, R.S.; Devitt, A. Redox regulation of protein damage in plasma. Redox Biol. 2014, 2, 430–435.
- Kelly, F.J.; Mudway, I.S. Protein oxidation at the air-lung interface. Amino Acids 2003, 25, 375–396.
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and uv radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559.
- Girotti, A.W. Mechanisms of lipid peroxidation. J. Free Radic. Biol. Med. 1985, 1, 87–95.
- Halliwell, B.; Gutteridge, J.M. Lipid peroxidation in brain homogenates: The role of iron and hydroxyl radicals. J. Neurochem. 1997, 69, 1330–1331.
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843.
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052.
- Sies, H.; Jones, D.P. Reactive oxygen species (ros) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Herb, M.; Gluschko, A.; Wiegmann, K.; Farid, A.; Wolf, A.; Utermohlen, O.; Krut, O.; Kronke, M.; Schramm, M. Mitochondrial reactive oxygen species enable proinflammatory signaling through disulfide linkage of nemo. Sci. Signal. 2019, 12.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. Ros and ros-mediated cellular signaling. Oxid Med. Cell Longev. 2016, 2016, 4350965.
- Marinho, H.S.; Cyrne, L.; Cadenas, E.; Antunes, F. The cellular steady-state of h2o2: Latency concepts and gradients. Methods Enzym. 2013, 527, 3–19.
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14.
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003.
- Antunes, F.; Cadenas, E. Estimation of h2o2 gradients across biomembranes. FEBS Lett. 2000, 475, 121–126.
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 101867.
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schroder, K.; Streckfuss-Bomeke, K.; Doroshow, J.H.; et al. Nox4 regulates insp3 receptor-dependent ca(2+) release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530.
- Wang, H.; Schoebel, S.; Schmitz, F.; Dong, H.; Hedfalk, K. Characterization of aquaporin-driven hydrogen peroxide transport. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183065.
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604.
- Herscovitch, M.; Comb, W.; Ennis, T.; Coleman, K.; Yong, S.; Armstead, B.; Kalaitzidis, D.; Chandani, S.; Gilmore, T.D. Intermolecular disulfide bond formation in the nemo dimer requires cys54 and cys347. Biochem. Biophys. Res. Commun. 2008, 367, 103–108.
- Jones, A.I.; Meshulam, T.; Oliveira, M.F.; Burritt, N.; Corkey, B.E. Extracellular redox regulation of intracellular reactive oxygen generation, mitochondrial function and lipid turnover in cultured human adipocytes. PLoS ONE 2016, 11, e0164011.
- Short, J.D.; Downs, K.; Tavakoli, S.; Asmis, R. Protein thiol redox signaling in monocytes and macrophages. Antioxid. Redox Signal. 2016, 25, 816–835.
- Rhee, S.G. Cell signaling. H2o2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883.
- Chiarugi, P.; Fiaschi, T.; Taddei, M.L.; Talini, D.; Giannoni, E.; Raugei, G.; Ramponi, G. Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet-derived growth factor receptor stimulation. J. Biol. Chem. 2001, 276, 33478–33487.
- Romero, L.C.; Aroca, M.A.; Laureano-Marin, A.M.; Moreno, I.; Garcia, I.; Gotor, C. Cysteine and cysteine-related signaling pathways in arabidopsis thaliana. Mol. Plant 2014, 7, 264–276.
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15.
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157.
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote tnfalpha-induced death and sustained jnk activation by inhibiting map kinase phosphatases. Cell 2005, 120, 649–661.
- Tonks, N.K. Redox redux: Revisiting ptps and the control of cell signaling. Cell 2005, 121, 667–670.
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 2002, 9, 387–399.
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1b in a431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372.
- Zhou, L.; Yeo, A.T.; Ballarano, C.; Weber, U.; Allen, K.N.; Gilmore, T.D.; Whitty, A. Disulfide-mediated stabilization of the ikappab kinase binding domain of nf-kappab essential modulator (nemo). Biochemistry 2014, 53, 7929–7944.
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421.
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686.
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561.
- Jung, S. Macrophages and monocytes in 2017: Macrophages and monocytes: Of tortoises and hares. Nat. Rev. Immunol. 2018, 18, 85–86.
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35.
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995.
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549.
- Haas, A. The phagosome: Compartment with a license to kill. Traffic 2007, 8, 311–330.
- Djaldetti, M.; Salman, H.; Bergman, M.; Djaldetti, R.; Bessler, H. Phagocytosis--the mighty weapon of the silent warriors. Microsc. Res. Tech. 2002, 57, 421–431.
- Schramm, M.; Wiegmann, K.; Schramm, S.; Gluschko, A.; Herb, M.; Utermohlen, O.; Kronke, M. Riboflavin (vitamin b2) deficiency impairs nadph oxidase 2 (nox2) priming and defense against listeria monocytogenes. Eur. J. Immunol. 2014, 44, 728–741.
- Mitchell, G.; Chen, C.; Portnoy, D.A. Strategies used by bacteria to grow in macrophages. Microbiol. Spectr. 2016, 4.
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203.
- Del Cerro-Vadillo, E.; Madrazo-Toca, F.; Carrasco-Marin, E.; Fernandez-Prieto, L.; Beck, C.; Leyva-Cobian, F.; Saftig, P.; Alvarez-Dominguez, C. Cutting edge: A novel nonoxidative phagosomal mechanism exerted by cathepsin-d controls listeria monocytogenes intracellular growth. J. Immunol. 2006, 176, 1321–1325.
- Aktan, F. Inos-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653.
- Utermohlen, O.; Karow, U.; Lohler, J.; Kronke, M. Severe impairment in early host defense against listeria monocytogenes in mice deficient in acid sphingomyelinase. J. Immunol. 2003, 170, 2621–2628.
- Alvarez-Dominguez, C.; Carrasco-Marin, E.; Lopez-Mato, P.; Leyva-Cobian, F. The contribution of both oxygen and nitrogen intermediates to the intracellular killing mechanisms of c1q-opsonized listeria monocytogenes by the macrophage-like ic-21 cell line. Immunology 2000, 101, 83–89.
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491.
- Allan, E.R.; Tailor, P.; Balce, D.R.; Pirzadeh, P.; McKenna, N.T.; Renaux, B.; Warren, A.L.; Jirik, F.R.; Yates, R.M. Nadph oxidase modifies patterns of mhc class ii-restricted epitopic repertoires through redox control of antigen processing. J. Immunol. 2014, 192, 4989–5001.
- Gluschko, A.; Herb, M.; Wiegmann, K.; Krut, O.; Neiss, W.F.; Utermohlen, O.; Kronke, M.; Schramm, M. The beta2 integrin mac-1 induces protective lc3-associated phagocytosis of listeria monocytogenes. Cell Host Microbe 2018, 23, 324–337.
- Craig, M.; Slauch, J.M. Phagocytic superoxide specifically damages an extracytoplasmic target to inhibit or kill salmonella. PLoS ONE 2009, 4, e4975.
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891.
- Slauch, J.M. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol. 2011, 80, 580–583.
- Yu, H.H.; Yang, Y.H.; Chiang, B.L. Chronic granulomatous disease: A comprehensive review. Clin. Rev. Allergy Immunol. 2020.
- Birmingham, C.L.; Canadien, V.; Kaniuk, N.A.; Steinberg, B.E.; Higgins, D.E.; Brumell, J.H. Listeriolysin o allows listeria monocytogenes replication in macrophage vacuoles. Nature 2008, 451, 350–354.
- Mitchell, G.; Cheng, M.I.; Chen, C.; Nguyen, B.N.; Whiteley, A.T.; Kianian, S.; Cox, J.S.; Green, D.R.; McDonald, K.L.; Portnoy, D.A. Listeria monocytogenes triggers noncanonical autophagy upon phagocytosis, but avoids subsequent growth-restricting xenophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E210–E217.
- Tattoli, I.; Sorbara, M.T.; Yang, C.; Tooze, S.A.; Philpott, D.J.; Girardin, S.E. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013, 32, 3066–3078.
- Heckmann, B.L.; Green, D.R. Lc3-associated phagocytosis at a glance. J. Cell Sci. 2019, 132.
- Herb, M.; Gluschko, A.; Schramm, M. Lc3-associated phagocytosis—The highway to hell for phagocytosed microbes. Semin. Cell Dev. Biol. 2020, 101, 68–76.
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257.
- Herb, M.; Gluschko, A.; Schramm, M. Lc3-associated phagocytosis initiated by integrin itgam-itgb2/mac-1 enhances immunity to listeria monocytogenes. Autophagy 2018, 14, 1462–1464.
- Charbonneau, M.E.; Passalacqua, K.D.; Hagen, S.E.; Showalter, H.D.; Wobus, C.E.; O’Riordan, M.X.D. Perturbation of ubiquitin homeostasis promotes macrophage oxidative defenses. Sci. Rep. 2019, 9, 10245.
- Noubade, R.; Wong, K.; Ota, N.; Rutz, S.; Eidenschenk, C.; Valdez, P.A.; Ding, J.; Peng, I.; Sebrell, A.; Caplazi, P.; et al. Nrros negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 2014, 509, 235–239.
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. Tlr signalling augments macrophage bactericidal activity through mitochondrial ros. Nature 2011, 472, 476–480.
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. Kinases mst1 and mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 2015, 16, 1142–1152.
- Fabrega, A.; Vila, J. Salmonella enterica serovar typhimurium skills to succeed in the host: Virulence and regulation. Clin. Microbiol. Rev. 2013, 26, 308–341.
- Eriksson, S.; Lucchini, S.; Thompson, A.; Rhen, M.; Hinton, J.C. Unravelling the biology of macrophage infection by gene expression profiling of intracellular salmonella enterica. Mol. Microbiol. 2003, 47, 103–118.
- Mastroeni, P.; Morgan, F.J.; McKinley, T.J.; Shawcroft, E.; Clare, S.; Maskell, D.J.; Grant, A.J. Enhanced virulence of salmonella enterica serovar typhimurium after passage through mice. Infect. Immun. 2011, 79, 636–643.
- Mastroeni, P.; Vazquez-Torres, A.; Fang, F.C.; Xu, Y.; Khan, S.; Hormaeche, C.E.; Dougan, G. Antimicrobial actions of the nadph phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. Ii. Effects on microbial proliferation and host survival in vivo. J. Exp. Med. 2000, 192, 237–248.
- Vazquez-Torres, A.; Xu, Y.; Jones-Carson, J.; Holden, D.W.; Lucia, S.M.; Dinauer, M.C.; Mastroeni, P.; Fang, F.C. Salmonella pathogenicity island 2-dependent evasion of the phagocyte nadph oxidase. Science 2000, 287, 1655–1658.
- De Groote, M.A.; Ochsner, U.A.; Shiloh, M.U.; Nathan, C.; McCord, J.M.; Dinauer, M.C.; Libby, S.J.; Vazquez-Torres, A.; Xu, Y.; Fang, F.C. Periplasmic superoxide dismutase protects salmonella from products of phagocyte nadph-oxidase and nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1997, 94, 13997–14001.
- Rhen, M. Salmonella and reactive oxygen species: A love-hate relationship. J. Innate Immun. 2019, 11, 216–226.
- Burton, N.A.; Schurmann, N.; Casse, O.; Steeb, A.K.; Claudi, B.; Zankl, J.; Schmidt, A.; Bumann, D. Disparate impact of oxidative host defenses determines the fate of salmonella during systemic infection in mice. Cell Host Microbe 2014, 15, 72–83.
- Fenlon, L.A.; Slauch, J.M. Phagocyte roulette in salmonella killing. Cell Host Microbe 2014, 15, 7–8.
- Hall, C.J.; Boyle, R.H.; Astin, J.W.; Flores, M.V.; Oehlers, S.H.; Sanderson, L.E.; Ellett, F.; Lieschke, G.J.; Crosier, K.E.; Crosier, P.S. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating beta-oxidation-dependent mitochondrial ros production. Cell Metab. 2013, 18, 265–278.
- Garaude, J.; Acin-Perez, R.; Martinez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villan, E.; Hervas-Stubbs, S.; Pelegrin, P.; Sander, L.E.; Enriquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045.
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific t cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236.
- Sazanov, L.A. The mechanism of coupling between electron transfer and proton translocation in respiratory complex i. J. Bioenerg. Biomembr. 2014, 46, 247–253.
- Orr, A.L.; Ashok, D.; Sarantos, M.R.; Shi, T.; Hughes, R.E.; Brand, M.D. Inhibitors of ros production by the ubiquinone-binding site of mitochondrial complex i identified by chemical screening. Free Radic. Biol. Med. 2013, 65, 1047–1059.
- Acin-Perez, R.; Iborra, S.; Marti-Mateos, Y.; Cook, E.C.L.; Conde-Garrosa, R.; Petcherski, A.; Munoz, M.D.M.; Martinez de Mena, R.; Krishnan, K.C.; Jimenez, C.; et al. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nat. Metab. 2020, 2, 974–988.
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012, 5, ra47.
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in nlrp3 inflammasome activation. Nature 2011, 469, 221–225.
- Webster, J.P. Dubey, j.P. Toxoplasmosis of animals and humans. Parasites Vectors 2010, 3, 112.
- Ybanez, R.H.D.; Ybanez, A.P.; Nishikawa, Y. Review on the current trends of toxoplasmosis serodiagnosis in humans. Front. Cell. Infect. Microbiol. 2020, 10, 204.
- Park, J.; Hunter, C.A. The role of macrophages in protective and pathological responses to toxoplasma gondii. Parasite Immunol. 2020, 42, e12712.
- Shrestha, S.P.; Tomita, T.; Weiss, L.M.; Orlofsky, A. Proliferation of toxoplasma gondii in inflammatory macrophages in vivo is associated with diminished oxygen radical production in the host cell. Int. J. Parasitol. 2006, 36, 433–441.
- Murray, H.W.; Rubin, B.Y.; Carriero, S.M.; Harris, A.M.; Jaffee, E.A. Human mononuclear phagocyte antiprotozoal mechanisms: Oxygen-dependent vs oxygen-independent activity against intracellular toxoplasma gondii. J. Immunol. 1985, 134, 1982–1988.
- Matta, S.K.; Patten, K.; Wang, Q.; Kim, B.H.; MacMicking, J.D.; Sibley, L.D. Nadph oxidase and guanylate binding protein 5 restrict survival of avirulent type iii strains of toxoplasma gondii in naive macrophages. mBio 2018, 9.
- Kim, J.H.; Lee, J.; Bae, S.J.; Kim, Y.; Park, B.J.; Choi, J.W.; Kwon, J.; Cha, G.H.; Yoo, H.J.; Jo, E.K.; et al. Nadph oxidase 4 is required for the generation of macrophage migration inhibitory factor and host defense against toxoplasma gondii infection. Sci. Rep. 2017, 7, 6361.
- Bedard, K.; Krause, K.H. The nox family of ros-generating nadph oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Laurindo, F.R.; Araujo, T.L.; Abrahao, T.B. Nox nadph oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775.
- Sciarretta, S.; Zhai, P.; Shao, D.; Zablocki, D.; Nagarajan, N.; Terada, L.S.; Volpe, M.; Sadoshima, J. Activation of nadph oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase rna-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ. Res. 2013, 113, 1253–1264.
- Martinez, A.; Prolo, C.; Estrada, D.; Rios, N.; Alvarez, M.N.; Pineyro, M.D.; Robello, C.; Radi, R.; Piacenza, L. Cytosolic fe-superoxide dismutase safeguards trypanosoma cruzi from macrophage-derived superoxide radical. Proc. Natl. Acad. Sci. USA 2019, 116, 8879–8888.
- Tomiotto-Pellissier, F.; Bortoleti, B.; Assolini, J.P.; Goncalves, M.D.; Carloto, A.C.M.; Miranda-Sapla, M.M.; Conchon-Costa, I.; Bordignon, J.; Pavanelli, W.R. Macrophage polarization in leishmaniasis: Broadening horizons. Front. Immunol. 2018, 9, 2529.
- Peters, N.C.; Sacks, D.L. The impact of vector-mediated neutrophil recruitment on cutaneous leishmaniasis. Cell Microbiol. 2009, 11, 1290–1296.
- Charmoy, M.; Auderset, F.; Allenbach, C.; Tacchini-Cottier, F. The prominent role of neutrophils during the initial phase of infection by leishmania parasites. J. Biomed. Biotechnol. 2010, 2010, 719361.
- Scott, P.; Novais, F.O. Cutaneous leishmaniasis: Immune responses in protection and pathogenesis. Nat. Rev. Immunol. 2016, 16, 581–592.
- Srivastava, S.; Shankar, P.; Mishra, J.; Singh, S. Possibilities and challenges for developing a successful vaccine for leishmaniasis. Parasites Vectors 2016, 9, 277.
- Matte, C.; Casgrain, P.A.; Seguin, O.; Moradin, N.; Hong, W.J.; Descoteaux, A. Leishmania major promastigotes evade lc3-associated phagocytosis through the action of gp63. PLoS Pathog. 2016, 12, e1005690.
- Alonso, D.; Serrano, E.; Bermejo, F.J.; Corral, R.S. Hif-1alpha-regulated mif activation and nox2-dependent ros generation promote leishmania amazonensis killing by macrophages under hypoxia. Cell. Immunol. 2019, 335, 15–21.
- To, E.E.; Vlahos, R.; Luong, R.; Halls, M.L.; Reading, P.C.; King, P.T.; Chan, C.; Drummond, G.R.; Sobey, C.G.; Broughton, B.R.S.; et al. Endosomal nox2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 2017, 8, 69.
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis c virus (hcv) proteins induce nadph oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to hcv-induced oxidative stress. J. Virol. 2009, 83, 12934–12946.
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by nadph oxidase 1. J. Virol. 2011, 85, 6795–6808.
- Vlahos, R.; Stambas, J.; Bozinovski, S.; Broughton, B.R.; Drummond, G.R.; Selemidis, S. Inhibition of nox2 oxidase activity ameliorates influenza a virus-induced lung inflammation. PLoS Pathog. 2011, 7, e1001271.
- Lang, P.A.; Xu, H.C.; Grusdat, M.; McIlwain, D.R.; Pandyra, A.A.; Harris, I.S.; Shaabani, N.; Honke, N.; Maney, S.K.; Lang, E.; et al. Reactive oxygen species delay control of lymphocytic choriomeningitis virus. Cell Death Differ. 2013, 20, 649–658.
- To, E.E.; Erlich, J.R.; Liong, F.; Luong, R.; Liong, S.; Esaq, F.; Oseghale, O.; Anthony, D.; McQualter, J.; Bozinovski, S.; et al. Mitochondrial reactive oxygen species contribute to pathological inflammation during influenza a virus infection in mice. Antioxid. Redox Signal. 2020, 32, 929–942.
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219.
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335.
- Dreux, M.; Chisari, F.V. Viruses and the autophagy machinery. Cell Cycle 2010, 9, 1295–1307.
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with hiv-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268.
- Sir, D.; Ou, J.H. Autophagy in viral replication and pathogenesis. Mol. Cells 2010, 29, 1–7.
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for sars-cov-2. Nat. Rev. Microbiol. 2020, 1–16.
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses hijack the lc3-i-positive edemosomes, er-derived vesicles exporting short-lived erad regulators, for replication. Cell Host Microbe 2010, 7, 500–508.
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347.
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and nf-kappab signaling. Cell Res. 2011, 21, 103–115.
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637.
- Roca, F.J.; Ramakrishnan, L. Tnf dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013, 153, 521–534.
- Hos, N.J.; Ganesan, R.; Gutierrez, S.; Hos, D.; Klimek, J.; Abdullah, Z.; Kronke, M.; Robinson, N. Type i interferon enhances necroptosis of salmonella typhimurium-infected macrophages by impairing antioxidative stress responses. J. Cell Biol. 2017, 216, 4107–4121.
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446.
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617.
- Li, T.; Kong, L.; Li, X.; Wu, S.; Attri, K.S.; Li, Y.; Gong, W.; Zhao, B.; Li, L.; Herring, L.E.; et al. Listeria monocytogenes upregulates mitochondrial calcium signalling to inhibit lc3-associated phagocytosis as a survival strategy. Nat. Microbiol. 2021.
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291.
- Zhao, Q.; Liu, J.; Deng, H.; Ma, R.; Liao, J.Y.; Liang, H.; Hu, J.; Li, J.; Guo, Z.; Cai, J.; et al. Targeting mitochondria-located circrna scar alleviates nash via reducing mros output. Cell 2020, 183, 76–93.e22.
- Lin, T.K.; Lin, K.J.; Lin, K.L.; Liou, C.W.; Chen, S.D.; Chuang, Y.C.; Wang, P.W.; Chuang, J.H.; Wang, T.J. When friendship turns sour: Effective communication between mitochondria and intracellular organelles in parkinson’s disease. Front. Cell Dev. Biol. 2020, 8, 607392.
- Maubach, G.; Schmadicke, A.C.; Naumann, M. Nemo links nuclear factor-kappab to human diseases. Trends Mol. Med. 2017, 23, 1138–1155.
- Hinz, M.; Scheidereit, C. The ikappab kinase complex in nf-kappab regulation and beyond. EMBO Rep. 2014, 15, 46–61.
- Herb, M.; Farid, A.; Gluschko, A.; Kronke, M.; Schramm, M. Highly efficient transfection of primary macrophages with in vitro transcribed mrna. J. Vis. Exp. 2019, 153, e60143.
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A. Metformin inhibits the production of reactive oxygen species from nadh:Ubiquinone oxidoreductase to limit induction of interleukin-1beta (il-1beta) and boosts interleukin-10 (il-10) in lipopolysaccharide (lps)-activated macrophages. J. Biol. Chem. 2015, 290, 20348–20359.
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656.
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596.
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in tnfr1-associated periodic syndrome (traps). J. Exp. Med. 2011, 208, 519–533.
- Rieber, N.; Hector, A.; Kuijpers, T.; Roos, D.; Hartl, D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin. Dev. Immunol. 2012, 2012, 252460.
- Deffert, C.; Carnesecchi, S.; Yuan, H.; Rougemont, A.L.; Kelkka, T.; Holmdahl, R.; Krause, K.H.; Schappi, M.G. Hyperinflammation of chronic granulomatous disease is abolished by nox2 reconstitution in macrophages and dendritic cells. J. Pathol. 2012, 228, 341–350.
- Whitmore, L.C.; Hilkin, B.M.; Goss, K.L.; Wahle, E.M.; Colaizy, T.T.; Boggiatto, P.M.; Varga, S.M.; Miller, F.J.; Moreland, J.G. Nox2 protects against prolonged inflammation, lung injury, and mortality following systemic insults. J. Innate Immun. 2013, 5, 565–580.
- Liang, S.; Ma, H.Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. Nadph oxidase 1 in liver macrophages promotes inflammation and tumor development in mice. Gastroenterology 2019, 156, 1156–1172.
- Liu, J.; Iwata, K.; Zhu, K.; Matsumoto, M.; Matsumoto, K.; Asaoka, N.; Zhang, X.; Ibi, M.; Katsuyama, M.; Tsutsui, M.; et al. Nox1/nadph oxidase in bone marrow-derived cells modulates intestinal barrier function. Free Radic. Biol. Med. 2020, 147, 90–101.
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2007, 27, 697–709.
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665.
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Dorfleutner, A.; Chu, L.; Stehlik, C. Inhibiting the inflammasome: One domain at a time. Immunol. Rev. 2015, 265, 205–216.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol. Cell 2002, 10, 417–426.
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome nlrs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735.
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. Ifi16 acts as a nuclear pathogen sensor to induce the inflammasome in response to kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375.
- Matsuoka, Y.; Yamashita, A.; Matsuda, M.; Kawai, K.; Sawa, T.; Amaya, F. Nlrp2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain 2019, 160, 2149–2160.
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel nlrp2 inflammasome. Glia 2013, 61, 1113–1121.
- Vladimer, G.I.; Weng, D.; Paquette, S.W.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The nlrp12 inflammasome recognizes yersinia pestis. Immunity 2012, 37, 96–107.
- Swanson, K.V.; Deng, M.; Ting, J.P. The nlrp3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarria-Smith, J.; Vance, R.E. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106.
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140.
- Van Bruggen, R.; Koker, M.Y.; Jansen, M.; van Houdt, M.; Roos, D.; Kuijpers, T.W.; van den Berg, T.K. Human nlrp3 inflammasome activation is nox1-4 independent. Blood 2010, 115, 5398–5400.
- Van de Veerdonk, F.L.; Smeekens, S.P.; Joosten, L.A.; Kullberg, B.J.; Dinarello, C.A.; van der Meer, J.W.; Netea, M.G. Reactive oxygen species-independent activation of the il-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc. Natl. Acad. Sci. USA 2010, 107, 3030–3033.
- Meissner, F.; Seger, R.A.; Moshous, D.; Fischer, A.; Reichenbach, J.; Zychlinsky, A. Inflammasome activation in nadph oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 2010, 116, 1570–1573.
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677.
- Dostert, C.; Guarda, G.; Romero, J.F.; Menu, P.; Gross, O.; Tardivel, A.; Suva, M.L.; Stehle, J.C.; Kopf, M.; Stamenkovic, I.; et al. Malarial hemozoin is a nalp3 inflammasome activating danger signal. PLoS ONE 2009, 4, e6510.
- Wingler, K.; Hermans, J.J.; Schiffers, P.; Moens, A.; Paul, M.; Schmidt, H.H. Nox1, 2, 4, 5: Counting out oxidative stress. Br. J. Pharm. 2011, 164, 866–883.
- Altenhofer, S.; Kleikers, P.W.; Radermacher, K.A.; Scheurer, P.; Rob Hermans, J.J.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H. The nox toolbox: Validating the role of nadph oxidases in physiology and disease. Cell Mol. Life Sci. 2012, 69, 2327–2343.
- Latz, E. Nox-free inflammasome activation. Blood 2010, 116, 1393–1394.
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabon, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. Nox4-dependent fatty acid oxidation promotes nlrp3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012.
- Naviaux, R.K. Oxidative shielding or oxidative stress? J. Pharmacol. Exp. Ther. 2012, 342, 608–618.
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290.
- Crane, D.D.; Bauler, T.J.; Wehrly, T.D.; Bosio, C.M. Mitochondrial ros potentiates indirect activation of the aim2 inflammasome. Front. Microbiol. 2014, 5, 438.
- Markvicheva, K.N.; Bilan, D.S.; Mishina, N.M.; Gorokhovatsky, A.Y.; Vinokurov, L.M.; Lukyanov, S.; Belousov, V.V. A genetically encoded sensor for h2o2 with expanded dynamic range. Bioorgan. Med. Chem. 2011, 19, 1079–1084.
- Gutscher, M.; Sobotta, M.C.; Wabnitz, G.H.; Ballikaya, S.; Meyer, A.J.; Samstag, Y.; Dick, T.P. Proximity-based protein thiol oxidation by h2o2-scavenging peroxidases. J. Biol. Chem. 2009, 284, 31532–31540.
- Bilan, D.S.; Pase, L.; Joosen, L.; Gorokhovatsky, A.Y.; Ermakova, Y.G.; Gadella, T.W.; Grabher, C.; Schultz, C.; Lukyanov, S.; Belousov, V.V. Hyper-3: A genetically encoded h(2)o(2) probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chem. Biol. 2013, 8, 535–542.
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286.
- Hernández-Barrera, A.; Quinto, C.; Johnson, E.A.; Wu, H.-M.; Cheung, A.Y.; Cárdenas, L. Chapter fifteen—Using hyper as a molecular probe to visualize hydrogen peroxide in living plant cells: A method with virtually unlimited potential in plant biology. In Methods in Enzymology; Cadenas, E., Packer, L., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 527, pp. 275–290.
- Ermakova, Y.G.; Bilan, D.S.; Matlashov, M.E.; Mishina, N.M.; Markvicheva, K.N.; Subach, O.M.; Subach, F.V.; Bogeski, I.; Hoth, M.; Enikolopov, G.; et al. Red fluorescent genetically encoded indicator for intracellular hydrogen peroxide. Nat. Commun. 2014, 5, 5222.
- Caldefie-Chezet, F.; Walrand, S.; Moinard, C.; Tridon, A.; Chassagne, J.; Vasson, M.P. Is the neutrophil reactive oxygen species production measured by luminol and lucigenin chemiluminescence intra or extracellular? Comparison with dcfh-da flow cytometry and cytochrome c reduction. Clin. Chim. Acta Int. J. Clin. Chem. 2002, 319, 9–17.
- Pavelkova, M.; Kubala, L. Luminol-, isoluminol- and lucigenin-enhanced chemiluminescence of rat blood phagocytes stimulated with different activators. Lumin. J. Biol. Chem. Lumin. 2004, 19, 37–42.
- Lundqvist, H.; Dahlgren, C. Isoluminol-enhanced chemiluminescence: A sensitive method to study the release of superoxide anion from human neutrophils. Free Radic. Biol. Med. 1996, 20, 785–792.
- Ushijima, Y.; Totsune, H.; Nishida, A.; Nakano, M. Chemiluminescence from human polymorphonuclear leukocytes activated with opsonized zymosan. Free Radic. Biol. Med. 1997, 22, 401–409.
- Li, Y.; Zhu, H.; Kuppusamy, P.; Roubaud, V.; Zweier, J.L.; Trush, M.A. Validation of lucigenin (bis-n-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J. Biol. Chem. 1998, 273, 2015–2023.
- Afanas’ev, I.B.; Ostrachovitch, E.A.; Korkina, L.G. Lucigenin is a mediator of cytochrome c reduction but not of superoxide production. Arch. Biochem. Biophys. 1999, 366, 267–274.
- Skatchkov, M.P.; Sperling, D.; Hink, U.; Mulsch, A.; Harrison, D.G.; Sindermann, I.; Meinertz, T.; Munzel, T. Validation of lucigenin as a chemiluminescent probe to monitor vascular superoxide as well as basal vascular nitric oxide production. Biochem. Biophys. Res. Commun. 1999, 254, 319–324.
- Hempel, S.L.; Buettner, G.R.; O’Malley, Y.Q.; Wessels, D.A.; Flaherty, D.M. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: Comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic. Biol. Med. 1999, 27, 146–159.
- Wang, Q.; Zou, M.H. Measurement of reactive oxygen species (ros) and mitochondrial ros in ampk knockout mice blood vessels. Methods Mol. Biol 2018, 1732, 507–517.
- Wolf, A.; Herb, M.; Schramm, M.; Langmann, T. The tspo-nox1 axis controls phagocyte-triggered pathological angiogenesis in the eye. Nat. Commun. 2020, 11, 2709.
- Dahlgren, C.; Karlsson, A. Respiratory burst in human neutrophils. J. Immunol. Methods 1999, 232, 3–14.
- Karakuzu, O.; Cruz, M.R.; Liu, Y.; Garsin, D.A. Amplex red assay for measuring hydrogen peroxide production from caenorhabditis elegans. Bio Protoc. 2019, 9, e3409.
- Mohanty, J.G.; Jaffe, J.S.; Schulman, E.S.; Raible, D.G. A highly sensitive fluorescent micro-assay of h2o2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J. Immunol. Methods 1997, 202, 133–141.
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brustle, A.; Itsumi, M.; et al. Glutathione primes t cell metabolism for inflammation. Immunity 2017, 46, 675–689.
- Mukhopadhyay, P.; Rajesh, M.; Hasko, G.; Hawkins, B.J.; Madesh, M.; Pacher, P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat. Protoc. 2007, 2, 2295–2301.
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043.
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.A.; Alshamsan, A. Mechanism of ros scavenging and antioxidant signalling by redox metallic and fullerene nanomaterials: Potential implications in ros associated degenerative disorders. Biochim. Biophys. Acta. Gen. Subj. 2017, 1861, 802–813.
- Poljsak, B.; Suput, D.; Milisav, I. Achieving the balance between ros and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell Longev. 2013, 2013, 956792.
- Gutteridge, J.M.; Halliwell, B. Antioxidants: Molecules, medicines, and myths. Biochem. Biophys. Res. Commun. 2010, 393, 561–564.
- Patriarca, S.; Furfaro, A.L.; Domenicotti, C.; Odetti, P.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N. Supplementation with n-acetylcysteine and taurine failed to restore glutathione content in liver of streptozotocin-induced diabetics rats but protected from oxidative stress. Biochim. Biophys. Acta 2005, 1741, 48–54.
- Zhou, J.; Coles, L.D.; Kartha, R.V.; Nash, N.; Mishra, U.; Lund, T.C.; Cloyd, J.C. Intravenous administration of stable-labeled n-acetylcysteine demonstrates an indirect mechanism for boosting glutathione and improving redox status. J. Pharm. Sci. 2015, 104, 2619–2626.
- Ezerina, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular h2s and sulfane sulfur production. Cell Chem. Biol. 2018, 25, 447–459.
- Bernardy, C.C.F.; Zarpelon, A.C.; Pinho-Ribeiro, F.A.; Calixto-Campos, C.; Carvalho, T.T.; Fattori, V.; Borghi, S.M.; Casagrande, R.; Verri, W.A., Jr. Tempol, a superoxide dismutase mimetic agent, inhibits superoxide anion-induced inflammatory pain in mice. Biomed. Res. Int. 2017, 2017, 9584819.
- Rak, R.; Chao, D.L.; Pluta, R.M.; Mitchell, J.B.; Oldfield, E.H.; Watson, J.C. Neuroprotection by the stable nitroxide tempol during reperfusion in a rat model of transient focal ischemia. J. Neurosurg. 2000, 92, 646–651.
- Haj-Yehia, A.I.; Nassar, T.; Assaf, P.; Nassar, H.; Anggard, E.E. Effects of the superoxide dismutase-mimic compound tempol on oxidant stress-mediated endothelial dysfunction. Antioxid. Redox Signal. 1999, 1, 221–232.
- Manzano, V.M.; Munoz, J.C.; Jimenez, J.R.; Puyol, M.R.; Puyol, D.R.; Kitamura, M.; Cazana, F.J. Human renal mesangial cells are a target for the anti-inflammatory action of 9-cis retinoic acid. Br. J. Pharm. 2000, 131, 1673–1683.
- Krishna, C.M.; Liebmann, J.E.; Kaufman, D.; DeGraff, W.; Hahn, S.M.; McMurry, T.; Mitchell, J.B.; Russo, A. The catecholic metal sequestering agent 1,2-dihydroxybenzene-3,5-disulfonate confers protection against oxidative cell damage. Arch. Biochem. Biophys. 1992, 294, 98–106.
- Hein, T.W.; Kuo, L. Ldls impair vasomotor function of the coronary microcirculation: Role of superoxide anions. Circ. Res. 1998, 83, 404–414.
- Dugas, A.J., Jr.; Castaneda-Acosta, J.; Bonin, G.C.; Price, K.L.; Fischer, N.H.; Winston, G.W. Evaluation of the total peroxyl radical-scavenging capacity of flavonoids: Structure-activity relationships. J. Nat. Prod. 2000, 63, 327–331.
- Davies, M.J.; Forni, L.G.; Willson, R.L. Vitamin e analogue trolox c. E.S.R. And pulse-radiolysis studies of free-radical reactions. Biochem. J. 1988, 255, 513–522.
- Matsushita, T.; Fukuda, K.; Yamamoto, H.; Yamazaki, K.; Tomiyama, T.; Oh, M.; Hamanishi, C. Effect of ebselen, a scavenger of reactive oxygen species, on chondrocyte metabolism. Mod. Rheumatol. 2004, 14, 25–30.
- Mugesh, G. Glutathione peroxidase activity of ebselen and its analogues: Some insights into the complex chemical mechanisms underlying the antioxidant activity. Curr. Chem. Biol. 2013, 7, 47–56.
- Nakamura, Y.; Feng, Q.; Kumagai, T.; Torikai, K.; Ohigashi, H.; Osawa, T.; Noguchi, N.; Niki, E.; Uchida, K. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant. Implication for inflammation-associated carcinogenesis. J. Biol. Chem. 2002, 277, 2687–2694.
- Frei, B.; England, L.; Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 6377–6381.
- Retsky, K.L.; Freeman, M.W.; Frei, B. Ascorbic acid oxidation product(s) protect human low density lipoprotein against atherogenic modification. Anti- rather than prooxidant activity of vitamin c in the presence of transition metal ions. J. Biol. Chem. 1993, 268, 1304–1309.
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457.
- Padh, H. Vitamin c: Newer insights into its biochemical functions. Nutr. Rev. 1991, 49, 65–70.
- Liang, W.J.; Johnson, D.; Jarvis, S.M. Vitamin c transport systems of mammalian cells. Mol. Membr. Biol. 2001, 18, 87–95.
- Hollander-Czytko, H.; Grabowski, J.; Sandorf, I.; Weckermann, K.; Weiler, E.W. Tocopherol content and activities of tyrosine aminotransferase and cystine lyase in arabidopsis under stress conditions. J. Plant Physiol. 2005, 162, 767–770.
- Ni, Y.; Eng, C. Vitamin e protects against lipid peroxidation and rescues tumorigenic phenotypes in cowden/cowden-like patient-derived lymphoblast cells with germline sdhx variants. Clin. Cancer Res. 2012, 18, 4954–4961.
- Newaz, M.A.; Nawal, N.N. Effect of alpha-tocopherol on lipid peroxidation and total antioxidant status in spontaneously hypertensive rats. Am. J. Hypertens. 1998, 11, 1480–1485.
- Niki, E. Oxidative stress and antioxidants: Distress or eustress? Arch. Biochem. Biophys. 2016, 595, 19–24.
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of nadph oxidase 1/4 inhibitors. Br. J. Pharm. 2017, 174, 1647–1669.
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of nadph oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2015, 23, 406–427.
- Wind, S.; Beuerlein, K.; Eucker, T.; Muller, H.; Scheurer, P.; Armitage, M.E.; Ho, H.; Schmidt, H.H.; Wingler, K. Comparative pharmacology of chemically distinct nadph oxidase inhibitors. Br. J. Pharm. 2010, 161, 885–898.
- Trevelin, S.C.; Dos Santos, C.X.; Ferreira, R.G.; de Sa Lima, L.; Silva, R.L.; Scavone, C.; Curi, R.; Alves-Filho, J.C.; Cunha, T.M.; Roxo-Junior, P.; et al. Apocynin and nox2 regulate nf-kappab by modifying thioredoxin-1 redox-state. Sci. Rep. 2016, 6, 34581.
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular nadph oxidases but an antioxidant. Hypertension 2008, 51(2), 211–217.
- Aldieri, E.; Riganti, C.; Polimeni, M.; Gazzano, E.; Lussiana, C.; Campia, I.; Ghigo, D. Classical inhibitors of nox nad(p)h oxidases are not specific. Curr. Drug Metab. 2008, 9, 686–696.
- Mora-Pale, M.; Weiwer, M.; Yu, J.; Linhardt, R.J.; Dordick, J.S. Inhibition of human vascular nadph oxidase by apocynin derived oligophenols. Bioorgan. Med. Chem. 2009, 17, 5146–5152.
- O’Donnell, B.V.; Tew, D.G.; Jones, O.T.; England, P.J. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil nadph oxidase. Biochem. J. 1993, 290 Pt 1, 41–49.
- Lambeth, J.D.; Krause, K.H.; Clark, R.A. Nox enzymes as novel targets for drug development. Semin. Immunopathol. 2008, 30, 339–363.
- Bloxham, D.P. The relationship of diphenyleneiodonium-induced hypoglycaemia to the specific covalent modification of nadh-ubiquinone oxidoreductase. Biochem. Soc. Trans. 1979, 7, 103–106.
- Geyer, O.; Podos, S.M.; Mittag, T. Nitric oxide synthase activity in tissues of the bovine eye. Graefe. Arch. Clin. Exp. Ophthalmol. Albrecht von Graefes Arch. Klin. Exp. Ophthalmol. 1997, 235, 786–793.
- Stuehr, D.J.; Fasehun, O.A.; Kwon, N.S.; Gross, S.S.; Gonzalez, J.A.; Levi, R.; Nathan, C.F. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1991, 5, 98–103.
- Tazzeo, T.; Worek, F.; Janssen, L. The nadph oxidase inhibitor diphenyleneiodonium is also a potent inhibitor of cholinesterases and the internal ca(2+) pump. Br. J. Pharm. 2009, 158, 790–796.
- Leusen, J.H.; Fluiter, K.; Hilarius, P.M.; Roos, D.; Verhoeven, A.J.; Bolscher, B.G. Interactions between the cytosolic components p47phox and p67phox of the human neutrophil nadph oxidase that are not required for activation in the cell-free system. J. Biol. Chem. 1995, 270, 11216–11221.
- Ten Freyhaus, H.; Huntgeburth, M.; Wingler, K.; Schnitker, J.; Baumer, A.T.; Vantler, M.; Bekhite, M.M.; Wartenberg, M.; Sauer, H.; Rosenkranz, S. Novel nox inhibitor vas2870 attenuates pdgf-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc. Res. 2006, 71, 331–341.
- Laleu, B.; Gaggini, F.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. First in class, potent, and orally bioavailable nadph oxidase isoform 4 (nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010, 53, 7715–7730.
- Strengert, M.; Jennings, R.; Davanture, S.; Hayes, P.; Gabriel, G.; Knaus, U.G. Mucosal reactive oxygen species are required for antiviral response: Role of duox in influenza a virus infection. Antioxid. Redox Signal. 2014, 20, 2695–2709.
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.; Burns, K.D.; Touyz, R.M.; et al. Critical role of nox4-based nadph oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010, 299, F1348–F1358.
- Gaggini, F.; Laleu, B.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. Design, synthesis and biological activity of original pyrazolo-pyrido-diazepine, -pyrazine and -oxazine dione derivatives as novel dual nox4/nox1 inhibitors. Bioorgan. Med. Chem. 2011, 19, 6989–6999.
- Aoyama, T.; Paik, Y.H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: Gkt137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327.
- Schildknecht, S.; Weber, A.; Gerding, H.R.; Pape, R.; Robotta, M.; Drescher, M.; Marquardt, A.; Daiber, A.; Ferger, B.; Leist, M. The nox1/4 inhibitor gkt136901 as selective and direct scavenger of peroxynitrite. Curr. Med. Chem. 2014, 21, 365–376.
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for nox4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619.
- Gorin, Y.; Cavaglieri, R.C.; Khazim, K.; Lee, D.Y.; Bruno, F.; Thakur, S.; Fanti, P.; Szyndralewiez, C.; Barnes, J.L.; Block, K.; et al. Targeting nadph oxidase with a novel dual nox1/nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am. J. Physiol. Ren. Physiol. 2015, 308, F1276–F1287.
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Metrailler-Ruchonnet, I.; Schappi, M.; Donati, Y.; Matthay, M.A.; Krause, K.H.; Barazzone Argiroffo, C. Nadph oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 972–981.
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The nox4 inhibitor gkt137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 718–726.
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology 2015, 149, 468–480.
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic investigations of the mitochondrial complex i inhibitor rotenone in the context of pharmacological and safety evaluation. Sci. Rep. 2017, 7, 45465.
- Stowe, D.F.; Camara, A.K. Mitochondrial reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid. Redox Signal. 2009, 11, 1373–1414.
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ros signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428.
- Palmer, G.; Horgan, D.J.; Tisdale, H.; Singer, T.P.; Beinert, H. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. Xiv. Location of the sites of inhibition of rotenone, barbiturates, and piericidin by means of electron paramagnetic resonance spectroscopy. J. Biol. Chem. 1968, 243, 844–847.
- Lambert, A.J.; Brand, M.D. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial nadh:Ubiquinone oxidoreductase (complex i). J. Biol. Chem. 2004, 279, 39414–39420.
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of parkinson disease: Multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035.
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790.
- Bleier, L.; Drose, S. Superoxide generation by complex iii: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331.
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex ii can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264.
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563.
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex iii. J. Biol. Chem. 2003, 278, 36027–36031.
- Muller, F.; Crofts, A.R.; Kramer, D.M. Multiple q-cycle bypass reactions at the qo site of the cytochrome bc1 complex. Biochemistry 2002, 41, 7866–7874.
- Starkov, A.A.; Fiskum, G. Myxothiazol induces h(2)o(2) production from mitochondrial respiratory chain. Biochem. Biophys. Res. Commun. 2001, 281, 645–650.

