 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | John Brewington | + 3937 word(s) | 3937 | 2021-04-27 06:01:02 | | | |
| 2 | Bruce Ren | Meta information modification | 3937 | 2021-05-08 04:43:19 | | |
Video Upload Options
The emergence of highly effective CFTR modulator therapy has led to significant improvements in health care for most patients with cystic fibrosis (CF). For some, however, these therapies remain inaccessible due to the rarity of their individual CFTR variants, or due to a lack of biologic activity of the available therapies for certain variants. One proposed method of addressing this gap is the use of primary human cell-based models, which allow preclinical therapeutic testing and physiologic assessment of relevant tissue at the individual level. Nasal cells represent one such tissue source and have emerged as a powerful model for individual disease study. The ex vivo culture of nasal cells has evolved over time, and modern nasal cell models are beginning to be utilized to predict patient outcomes.
1. Introduction
Cystic fibrosis (CF) is a multisystem, autosomal recessive disease characterized by progressive obstructive respiratory failure [1]. Mutations in the gene encoding the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) protein cause CF, with >2000 variants described to date, though not all variants result in CF [2][3]. This protein acts as an ion channel at the epithelial surface, responsible for gated passage of chloride and bicarbonate [4][5][6]. Variants in the CFTR gene result in several forms of protein dysfunction or deficiency. For example, the most common variant, F508del CFTR, results primarily in improper protein trafficking to the cell surface, which is necessary for protein function [7]. Other variants may result in reduced protein production, altered channel conductance properties, impaired channel gating, or more. Some of these variants have been studied and grouped into classification schemes of the resultant protein problem, though the impacts of many CFTR variants remain unknown [8].
Understanding the protein impact of common CFTR variants required concerted efforts across many groups and was largely informed by studies of heterologous models over-expressing a single mutant allele [9][10][11][12][13]. These efforts have paid dividends, leading to the creation of small-molecule therapeutics that improve various forms of protein dysfunction [14][15][16][17][18]. Termed “CFTR Modulators”, these drugs have altered the landscape of CF care, shifting from a model of symptom management to one of symptom prevention, treating the protein-level cause of the disease. Highly effective modulator therapies are now available to >85% of patients in North America with CF, and in Phase 3 trials have produced significant improvements in lung function, nutritional status, and patient-reported outcome measures [14][15].
Despite these remarkable accomplishments, several key issues remain. First, modulator therapy is not available to all patients with CF. Some carry two alleles that will not be amenable to small-molecule correction, such as those with two nonsense variants. Others, however, carry alleles that may respond to such therapies, but have not been evaluated due to the rarity of the variant. It has been estimated that approximately half of the >2000 identified CFTR variants are carried by 5 individuals in the world or less; these individuals have been, by nature of their rarity, excluded from both clinical trials and heterologous model-driven studies of CFTR biology [10][19][20]. It is particularly noteworthy that such rare and ultra-rare variants are more likely to be present in individuals in minority racial groups, further exacerbating existing disparities in care compared to their Caucasian counterparts [21]. A second issue with the current therapeutic environment is the limited understanding of phenotypic and therapeutic variance in CF. Though modulators are prescribed based upon genotype, prior studies have demonstrated that >50% of disease variance is unrelated to this genetic factor [22]. This disease variance has been re-demonstrated across modulator trials, with wide variation in patient outcomes [14][15][16][17][18].
One proposed way to address these issues is through the study of patient-derived laboratory models generated from disease-relevant tissue sources. Through analysis of patient-specific laboratory outcomes and linked clinical data, the cellular mechanisms underlying disease and therapeutic variance may be elucidated. Using the same models, analysis of CFTR function with various treatments, such as modulators, may provide a preclinical assessment of patient response, and therefore be a useful tool for matching patients with the optimal treatments. This patient-treatment matching has been termed “theratyping,” or classifying a patient’s CFTR variants by the efficacy of treatments for those variants [23]. Some have approached such theratyping work in heterologous models, including Fisher Rat Thyroid cells; this approach has been successful in pushing FDA expansion of modulator label indications [24]. This approach, while powerful, is limited to more common variants, and the heterologous model data has not always held true to the human condition [25][26][27]. Using patient-derived models for theratyping efforts, conversely, will theoretically be of greater relevance to the individual subject, representing not only both of their CFTR alleles, but also any other genetic or cellular determinants of disease. To be clear, both heterologous and patient-derived models have important roles, strengths, and weakness with relation to optimizing and expanding CFTR-focused therapeutics. For the present review, however, focus will be placed on patient-specific models.
Use of patient-specific models to study CF is not a novel concept. Bronchial epithelial cells grown at air–liquid interface (ALI), for example, have been instrumental in constructing the current understanding of CFTR biology in the airway [28]. These models, however, are highly invasive to acquire (requiring lung transplant), and therefore are not well-suited to patient-specific study [29]. Seeking less invasive sources, many investigators have capitalized on other disease-relevant tissues, with extensive research in intestinal and nasal cell-based models [30][31][32][33]. Intestinal models are frequently used across Europe with great success, and have demonstrated robust growth capacity, clear disease relevance, and promise as a platform for therapeutic development [33]. These models, however, have not gained as much traction in North America, with a limited number of centers performing these assays regularly [23]. Additionally, while the intestine is a critically important tissue in CF disease, the mortality of the disease stems from the respiratory tract. As ion transport in the airway and the gut are not equivalent, a respiratory model may carry benefits over an intestinal source.
2. Historical Nasal Cell Models
Conceptually, the use of nasal models to understand the functional role of ion transport in CF may have started with the description of functional, in vivo differences in nasal potential difference between CF and non-CF subjects in 1981 [34]. Subsequent studies using this assay demonstrated disease-relevant properties of the nasal mucosa, such as increased sodium transport and decreased chloride efflux in subjects with CF, which had been previously described in difficult-to-obtain lower airway samples [35]. Transitioning these in vivo studies to in vitro assays to better understand the cellular mechanisms of such observations was a logical step, but impeded by a lack of reliable culture methodology. One early method, for example, was the repopulated heterologous graft technique, in which human nasal cells were dissociated from polyp samples, seeded onto denuded murine tracheal explants, and then implanted into immune-deficient mice [36]. After several weeks, these tracheal implants were collected and assayed for ion flux. This system generated useful results, but viability and repeatability were limited, with early reports of culture success well under 50% [36].
A simpler system for culturing nasal cells became more popular in the mid-1980s, relying on suspension culture using serum-supplemented media. An early description of this technique demonstrated excellent success rates for short-term cultures of polyp tissue, and utilized this model to characterize deficient chloride uptake in these nasal cells [37]. Importantly, however, phenotypic analysis of the cultured cells did not reveal a classic respiratory morphology, lacking the columnar shape and organization seen in non-dissociated samples [37]. Nonetheless, these submerged, incompletely differentiated cultures became quite useful in describing the physiologic impact of CFTR deficiency on the airway epithelium. Through the use of chloride uptake/efflux assays, patch-clamp, and more, these studies demonstrated altered ion transport, increased transmembrane potential difference, increased sodium permeability, and reduced chloride permeability in samples from those with CF, all now hallmarks of the CF respiratory epithelium [37][38][39][40][41]. Similar cultures were utilized through the late 1990s to study respiratory ciliogenesis, CF-relevant host-pathogen interactions (e.g., Pseudomonas aeruginosa, Staphylococcus aureus), gene therapy, and early F508del protein correction with therapeutics [42][43][44][45][46][47][48].
In this submerged culture system, two key limitations reduced both throughput and applicability. Submerged cultures of nasal cells did not fully recapitulate the mature respiratory epithelium, with impaired morphologic differentiation [37]. This lack of differentiation may reduce the in vivo disease relevance, especially in studies where mature tight junction and barrier formation is essential (e.g., gene therapy). The ability to repeat such studies with high numbers was also limited by poor replication of nasal cells in vitro. Unlike other tissue sources with high growth capacity (such as tumor cells), overall expansion of nasal cells using traditional methods is limited to a few passages. In order to overcome this, investigators primarily utilized tissue sources with high cell yield; specifically, polyp and surgical specimens [37][41]. This drastically reduced the potential subject recruitment pool to a convenience sample of those undergoing surgical procedures, largely eliminating pediatric subjects from consideration. Attempts were made at culture using samples obtained by cytology brushing but had limited success. In one such early brushed nasal cell study, while numerous endpoints were achieved, only 7 of 17 cultures reached confluence for full study, highlighting the limited expansion potential of HNEs [49].
A number of methods were trialed to overcome this limitation. Immortalization of HNE cultures was feasible and successful, but required resource commitment, and the long-term relevance of the cultures was unclear [50]. Several modifications of the early culture media, such as altering the serum component or adding feeder fibroblasts, allowed for improved, but still suboptimal expansion [51][52]. Ultimately, however, it was the emergence of conditional reprogramming culture (CRC) technologies in 2012 that allowed for more widespread use of HNEs for CF study [53]. This method utilizes media containing a Rho kinase inhibitor (Y-27632) and irradiated feeder fibroblasts to drastically improve cell growth across a number of tissue sources [53]. In nasal and lower airway cells, the use of CRC methodology has been reported to allow >300-fold expansion of the initial cell sample compared to traditional methods through enriching a progenitor population and preserving the basal cell characteristics of the culture, potentially via a KLF11-dependent action [54][55]. This approach has empowered brushed HNE cell studies from infants up to adults, bypassing the need for surgical samples [56][57].
Novel methods of increasing cell expansion have also emerged, such as dual-SMAD inhibition, but are not yet well characterized [58]. Prior to routine adoption of these methods, additional studies will be required to optimize protocols and define differences and similarities between methods. Regardless, numerous HNE studies now utilize CRC or other expansion methodology to allow for improved cell growth, followed by culture into a differentiated model such as ALI or spheroid cultures. These well-differentiated HNE cultures, patterned after success with bronchial or intestinal models, continue to increase the disease relevance of cultured HNE studies by recapitulating the native airway morphology.
3. Air–Liquid Interface Cultures
Primary human bronchial epithelial (HBE) tissue planar cultures grown at ALI have been instrumental in therapeutics development for cystic fibrosis and have played an important translational role in leading to the FDA approval of the current modulators. HBE planar cultures remain the gold standard in human tissue-based respiratory models for CF study [23]. The traditional source of tissue for these cultures is explanted lung tissue obtained during lung transplantation and therefore is quite rare, resulting in limited utility for theratyping. The need for a more readily available respiratory tissue source led to the development of HBE cultures from cells obtained during sedated bronchoscopic procedures, and eventually to HNE cultures grown at ALI with cells obtained from nasal brushing [28][59][60]. The application to theratyping is thus drastically improved with minimally invasive tissue collection which even allows for recollection in instances of contamination or failed culture growth.
First described in the late 1980s, culture at ALI allows for the growth of well-differentiated pseudostratified mucociliary epithelium with important physiologic characteristics of in vivo airway epithelium [28][60][61]. In this method, cells are plated onto suspended, semi-porous membranes, with media below the membrane and air above the cells [60]. While prior attempts at primary airway culture grown submerged on plastic dishes resulted in a poorly differentiated squamous morphology, it was found that cellular polarization enabled by the ALI orientation proved critical to mucociliary differentiation [60]. Long-term culture growth over numerous subsequent passages was enabled by the use of fibroblast feeder layers to stimulate proliferation [59].
Electrophysiological assessment of HBE planar cultures grown at ALI was critical in advancing the biologic understanding of CF. In this assay, short circuit current and transepithelial voltage across the polarized membrane is measured in Ussing chambers [62][63]. A series of solutions is applied to the culture to isolate and stimulate CFTR so that ion efflux through CFTR can be directly measured. This technique allows for the detection of CFTR activity and assessment of alterations in function provoked by modulators or other substances, and has been critical in modulator development and expansion [13][24][64][65]. In addition to electrophysiologic assessment, evaluations of ciliary function, inflammatory response, and airway surface liquid physiology have been described using cultures grown in ALI [66][67][68][69].
To adapt these techniques to nasal cells, brushed and expanded nasal cells are directly seeded onto porous supports. Media, which varies by lab, maintains the culture, promotes differentiation and expansion, and is exchanged regularly [31][70]. Depending on the media used, HNE ALI cultures mature within 4–6 weeks [31][70][71]. The HNE culture recapitulation of lower airway epithelial morphology and physiology allows for HNE use in theratyping as well as in tissue-specific physiologic studies; immunofluorescence demonstrating such morphologic characteristics can be found in Figure 1.
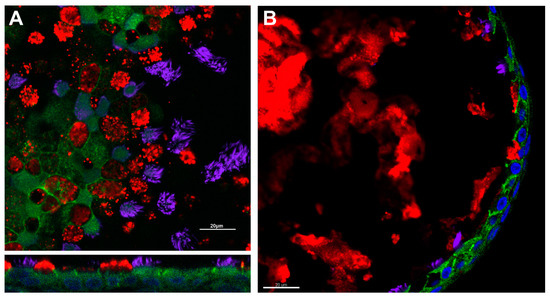
Figure 1. Respiratory epithelial characteristics of HNE models. Immunofluorescence demonstrates the presence of adherens junctions (e-cadherin, green), cilia (acetylated alpha-tubulin, purple), and mucus production (muc5ac, red) in air–liquid interface (Panel (A)) and spheroid cultures (Panel (B)). Nuclei are highlighted in blue by DAPI. Scale bar = 20 µm. Image courtesy of Brewington laboratory.
Electrophysiologic assessment of CFTR activity in HNE ALI cultures has the capacity to discriminate between CF, WT, and CF disease with partial CFTR function, and modulator corrected mutant CFTR [72][73]. Among subjects with CF, modulated, ex vivo CFTR activity in HNE cells has been shown to correlate with clinical improvements ppFEV1 and sweat chloride, both at the individual level and against historical trial cohort data [32][70][71][72][74][75]. While Ussing chamber studies represent the gold standard in electrophysiological assessment of modulator-rescued CFTR function they are limited by the need for highly specialized equipment and a time and labor intensive process, resulting in a low-throughput assay. A medium-throughput assay for profiling of CFTR modulator efficacy that uses fluorescence-based identification of chloride ion conduction has been described in HNE with good congruence to Ussing chamber data in showing individual patient response to modulators [76].
Despite the rapidly emerging evidence, the use of HNE planar cultures grown at ALI remains limited by uncertainty regarding the relevance of nasal cell culture to the lower airways, specifically in disease states such as CF that have profound lower airways pathology. There is additional uncertainty regarding the relevance of the in vitro environment to the actual human nasal or respiratory epithelium. While the studies above have linked nasal cells to individual or unrelated cohort patient studies, the numbers in these comparisons are small, and the precision of this linkage remains unclear [70][71][72][74][75]. Primary human nasal cell cultures are limited by the number of times they can be passaged as well as by a risk for contamination as compared to immortalized cell lines, though the relevance of primary culture likely offsets this downside for human studies. Notably, cells for nasal tissue culture are more accessible than bronchial or intestinal cells and can be obtained with minimal discomfort to the patient.
4. Three-Dimensional Cultures
In contrast to ALI cultures, several three-dimensional nasal cell culture models have been described for CF study. Many, though not all, of these models meet the generally held definition of an “organoid,” including three-dimensional structure recapitulating the in vivo tissue coupled with recreation of a tissue-specific function (e.g., mucus production, ciliary motility). Given the relative simplicity of these HNE models, however, most have utilized the term “spheroids” or “nasospheroids” [57][77][78]. For this review, we will utilize the unifying term “spheroids,” but readily acknowledge the laxity of terminology. Regardless of nomenclature, these models all share a three-dimensional, cystic form in which epithelial cells reliably polarize and recreate physiologic characteristics of the respiratory epithelium.
Early descriptions of HNE spheroids from the 1990s largely focus on a submersion culture method. In these models, brushed HNE cells or dissociated nasal polyp tissue is plated in culture flasks or dishes, then manually moved either by hand or on a shaker for the first days of growth [77][79]. This mechanical disruption prevents cell adhesion to the culture vessel, encouraging instead the formation of spherical clusters of epithelial cells with a clear inner lumen, and epithelial cells polarized with the apical surface to the outside. These spheroids morphologically resemble the intact airway epithelium, and provided a model for studies of fluid transport, mucociliary clearance, and ciliogenesis [77][79][80][81][82]. In addition to these physiologic studies, these apex-out, floating spheroids were studied using microelectrodes to characterize ion transport across the nasal mucosa [83]. This study demonstrated clear differences between CF and non-CF samples, establishing the capacity of nasal spheroids to discriminate incremental CFTR function [83].
More recently, investigators have adapted methodology utilized for intestinal organoid studies in CF to nasal spheroids. In this approach, expanded HNEs are embedded in a three-dimensional matrix (typically Matrigel, Corning, NY, USA), which supports the spherical organization of the cells without the need for mechanical agitation [30][57][78][84]. Much like previous spheroid models, these cultures recapitulate the mature respiratory epithelium, including ciliation, mucus production, and formation of cell-to-cell junctions [57][78]. Immunofluorescence demonstrating these morphologic characteristics can be found in Figure 1. By altering the matrix density, cultures can be generated with the epithelial cell apex to the inside/lumen of the spheroid or to the outside [57][78]. To quantify CFTR function, ion transport is stimulated through cAMP-dependent pathways and spheroid size is monitored over time [30][57][78][84]. Using this approach, several groups have reported the capacity of these spheroids to discriminate CF cultures from non-CF, and to demonstrate pharmacologic rescue of CFTR using modulator compounds [57][78][84].
There are several potential benefits to these three-dimensional models over ALI cultures, as well as limitations. After an expansion phase, these cultures reach maturity and readiness for study within 1–2 weeks, typically twice as quickly as ALI cultures [57]. In addition, this methodology can generate hundreds of spheroids, drastically increasing the testing and analysis possibilities over a relatively small number of ALI cultures [30][78]. The analysis of these cultures can be partially automated, increasing throughput [84]. Moreover, by analyzing fluid efflux, these models provide a complimentary readout that may have more direct physiologic relevance to the CF disease state compared to measurements of ion transport in ALI cultures.
Conversely, these spheroid cultures carry limitations relative to ALI cultures. For one, the model variance in spheroids has been reported to be greater than that typically associated with ALI cultures. This is partially offset by increasing the number of measurements but must be considered in study design [57][78]. In addition, the relative novelty of this approach requires robust study to link model results to clinical or established laboratory data before relevance can be established. These efforts are limited by a lack of shared and optimized SOPs, reducing the capacity to compare studies between labs. Due to these limitations, HNE spheroid cultures have, thus far, been less utilized compared to ALI cultures.
5. Relevance of Nasal Cell Models to the Lower Airways
Establishing the relevance of nasal cell models to the lower airways is of obvious importance to any attempt to use these models for theratyping, personalized studies, or more broadly as surrogates for the study of lower airways diseases. There is a growing body of evidence that suggests that there is sufficient similarity between the morphology and function of nasal cell models and bronchial epithelial tissue to allow HNE to be used broadly as surrogates for HBE in future research in CF and other lower airways disease [31][32][73][85][86]. Several studies have already reported correlations between the results of studies in nasal cell culture models and meaningful clinical outcomes [70][71][72][74][75]. However, it is important to note that there may be specific situations in which HNE cultures would not be suitable for use as a surrogate for bronchial epithelium and the criteria to determine their suitability is ultimately contingent upon the exact question being asked.
Characterization of the structure, morphology, and cellular composition of HNE planar cultures has demonstrated the recapitulation of specific characteristics of mature respiratory epithelia [31][66]. Submerged nasal epithelial cultures are visually indistinguishable in their morphology from submerged bronchial epithelial cultures, have similar receptor expression and, although they differ in absolute mediator levels, both show similar response to cytokine stimulation [85]. Submerged cell culture models, however, are undifferentiated and have mostly been supplanted by cultures grown at ALI as they appear to have greater fidelity to in vivo conditions. HNE cultures grown at ALI display pseudostratified columnar epithelial morphology with a cellular composition that consists of ciliated cells, mucous producing cells, basal cells, and ionocytes [31][66][87]. The number of ionocytes in nasal epithelial cultures and bronchial cultures is significantly different although CFTR function is not, and it is thought that the difference reflects a proximal to distal gradient in ionocyte numbers within the respiratory tract [87]. The presence of tight-junctional proteins has been confirmed in multiple studies and contributes to epithelial barrier integrity within HNE planar cultures at ALI [31][66].
Physiologically, HNE cultures have been shown to recapitulate key components of HBE cultures that are critical to the analysis of airways diseases. For example, electron microscopic examination has shown that the cilia in fully differentiated HNE planar cultures grown at ALI have a typical ultrastructure with normal axonemal microtubule and dynein arm arrangement as well as intact ciliary beat [66]. This allows for the evaluation of ciliopathies in the absence of possible confounding “secondary” dysfunction of cilia that can occur in fresh primary cells upon harvesting [67]. Of particular interest in CF, electrophysiologic assays have demonstrated the presence of functional CFTR in HNE cultures, and paired samples of brushed HNE and brushed HBE from the same patient (among a cohort of patients with CF) have shown similar CFTR dependent ion transport and a strong correlation in the ability to detect CFTR response to modulation between the two culture types [31].
In other disease states direct comparisons between HNE and HBE cultures have supported the use of HNE cultures as surrogates for HBE cultures with some limitations. In asthmatic patients the physiologic response to IL-13 stimulus is similar between HNE and HBE cultures at ALI, but there are distinct morphologic differences with respect to the number of goblet and ciliated cells in the nasal epithelial cultures compared to the bronchial epithelial cultures [88]. This may reflect an inherent restriction of goblet cell hyperplasia to the lower airways in asthma but is an important consideration in terms of model selection. Nasal cell cultures have been used in the study of viral entry, receptors and internalization, viral replication and innate immune responses [89]. Studies using paired nasal and bronchial cell cultures have shown similar antiviral and viral induced pro-inflammatory responses between to the two tissue sources [90]. Nasal gene expression profiling has the ability to differentiate patients with COPD from patients without COPD, and analysis of the nasal versus bronchial COPD gene expression profile shows significant overlap between the epithelia [91]. Comparisons of the inflammatory response in paired HNE and HBE cultures at ALI from patients with COPD shows correlation in IL-8 response to pseudomonas aeruginosa lipopolysaccharide between the culture types but differing IL-6 response, as well as differential response to cigarette spoke exposure in Toll-like receptor 4 expression [92].
Taken together, these studies demonstrate overall similarities in structural and functional characteristics of HNE and HBE cultures at ALI, though subtle differences exist. While differences in inflammatory mediators or genomic profiling may prompt hesitancy for certain investigations, functional analyses of CFTR activity have consistently supported HNEs as an adequate surrogate for HBE study [30][70][71].
References
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001.
- The Clinical and Functional TRanslation of CFTR (CFTR2). Copyright 2011 US CF Foundation, Johns Hopkins University, The Hospital for Sick Children. Available online: (accessed on 12 April 2021).
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073.
- Smith, J.J.; Welsh, M.J. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J. Clin. Investig. 1992, 89, 1148–1153.
- Anderson, M.P.; Gregory, R.J.; Thompson, S.; Souza, D.W.; Paul, S.; Mulligan, R.C.; Smith, A.E.; Welsh, M.J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991, 253, 202–205.
- Bear, C.E.; Li, C.H.; Kartner, N.; Bridges, R.J.; Jensen, T.J.; Ramjeesingh, M.; Riordan, J.R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 1992, 68, 809–818.
- Du, K.; Sharma, M.; Lukacs, G.L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25.
- Palmer, L.B.; Smaldone, G.C. Reduction of bacterial resistance with inhaled antibiotics in the intensive care unit. Am. J. Respir. Crit. Care Med. 2014, 189, 1225–1233.
- Dérand, R.; Bulteau-Pignoux, L.; Mettey, Y.; Zegarra-Moran, O.; Howell, L.D.; Randak, C.; Galietta, L.J.; Cohn, J.A.; Norez, C.; Romio, L.; et al. Activation of G551D CFTR channel with MPB-91: Regulation by ATPase activity and phosphorylation. Am. J. Physiol. Cell Physiol. 2001, 281, C1657–C1666.
- Rowe, S.M.; Pyle, L.C.; Jurkevante, A.; Varga, K.; Collawn, J.; Sloane, P.A.; Woodworth, B.; Mazur, M.; Fulton, J.; Fan, L.; et al. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm. Pharmacol. Ther. 2010, 23, 268–278.
- Suaud, L.; Carattino, M.; Kleyman, T.R.; Rubenstein, R.C. Genistein improves regulatory interactions between G551D-cystic fibrosis transmembrane conductance regulator and the epithelial sodium channel in Xenopus oocytes. J. Biol. Chem. 2002, 277, 50341–50347.
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P., Jr.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245.
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848.
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948.
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819.
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784.
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672.
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023.
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.T.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1116–1126.
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013, 45, 1160–1167.
- McGarry, M.E.; McColley, S.A. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr. Pulmonol. 2021.
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56.
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34.
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830.
- Fidler, M.C.; Buckley, A.; Sullivan, J.C.; Statia, M.; Boj, S.F.; Vries, R.G.J.; Munck, A.; Higgins, M.; Moretto Zita, M.; Negulescu, P.; et al. G970R-CFTR Mutation (c.2908G>C) Results Predominantly in a Splicing Defect. Clin. Transl. Sci. 2020.
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; De Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748.
- Laselva, O.; Moraes, T.J.; He, G.; Bartlett, C.; Szàrics, I.; Ouyang, H.; Gunawardena, T.N.A.; Strug, L.; Bear, C.E.; Gonska, T. The CFTR Mutation c.3453G > C (D1152H) Confers an Anion Selectivity Defect in Primary Airway Tissue that Can Be Rescued by Ivacaftor. J. Pers. Med. 2020, 10, 40.
- Fulcher, M.L.; Randell, S.H. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol. Biol. 2013, 945, 109–121.
- Randell, S.H.; Fulcher, M.L.; O’Neal, W.; Olsen, J.C. Primary epithelial cell models for cystic fibrosis research. Methods Mol. Biol. 2011, 742, 285–310.
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J., 3rd; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Generation of Human Nasal Epithelial Cell Spheroids for Individualized Cystic Fibrosis Transmembrane Conductance Regulator Study. J. Vis. Exp. 2018.
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J., 3rd; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018, 3.
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126.
- de Poel, E.; Lefferts, J.W.; Beekman, J.M. Intestinal organoids for Cystic Fibrosis research. J. Cyst. Fibros. 2020, 19 (Suppl. 1), S60–S64.
- Knowles, M.; Gatzy, J.; Boucher, R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N. Engl. J. Med. 1981, 305, 1489–1495.
- Knowles, M.; Gatzy, J.; Boucher, R. Relative ion permeability of normal and cystic fibrosis nasal epithelium. J. Clin. Investig. 1983, 71, 1410–1417.
- Yankaskas, J.R.; Knowles, M.R.; Gatzy, J.T.; Boucher, R.C. Persistence of abnormal chloride ion permeability in cystic fibrosis nasal epithelial cells in heterologous culture. Lancet 1985, 1, 954–956.
- Stutts, M.J.; Cotton, C.U.; Yankaskas, J.R.; Cheng, E.; Knowles, M.R.; Gatzy, J.T.; Boucher, R.C. Chloride uptake into cultured airway epithelial cells from cystic fibrosis patients and normal individuals. Proc. Natl. Acad. Sci. USA 1985, 82, 6677–6681.
- Murphy, E.; Cheng, E.; Yankaskas, J.; Stutts, M.J.; Boucher, R.C. Cell calcium levels of normal and cystic fibrosis nasal epithelium. Pediatr. Res. 1988, 24, 79–84.
- Boucher, R.C.; Cotton, C.U.; Gatzy, J.T.; Knowles, M.R.; Yankaskas, J.R. Evidence for reduced Cl- and increased Na+ permeability in cystic fibrosis human primary cell cultures. J. Physiol. 1988, 405, 77–103.
- Kunzelmann, K.; Kathöfer, S.; Greger, R. Na+ and Cl- conductances in airway epithelial cells: Increased Na+ conductance in cystic fibrosis. Pflug. Arch. 1995, 431, 1–9.
- Prulière-Escabasse, V.; Fanen, P.; Dazy, A.C.; Lechapt-Zalcman, E.; Rideau, D.; Edelman, A.; Escudier, E.; Coste, A. TGF-beta 1 downregulates CFTR expression and function in nasal polyps of non-CF patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L77–L83.
- Jorissen, M.; Van der Schueren, B.; Van den Berghe, H.; Cassiman, J.J. In vitro ciliogenesis in respiratory epithelium of cystic fibrosis patients. Ann. Otol. Rhinol. Laryngol. 1991, 100, 366–371.
- Plotkowski, M.C.; Chevillard, M.; Pierrot, D.; Altemayer, D.; Puchelle, E. Epithelial respiratory cells from cystic fibrosis patients do not possess specific Pseudomonas aeruginosa-adhesive properties. J. Med. Microbiol. 1992, 36, 104–111.
- Bajolet-Laudinat, O.; Girod-de Bentzmann, S.; Tournier, J.M.; Madoulet, C.; Plotkowski, M.C.; Chippaux, C.; Puchelle, E. Cytotoxicity of Pseudomonas aeruginosa internal lectin PA-I to respiratory epithelial cells in primary culture. Infect. Immun. 1994, 62, 4481–4487.
- Ulrich, M.; Herbert, S.; Berger, J.; Bellon, G.; Louis, D.; Münker, G.; Döring, G. Localization of Staphylococcus aureus in infected airways of patients with cystic fibrosis and in a cell culture model of S. aureus adherence. Am. J. Respir. Cell Mol. Biol. 1998, 19, 83–91.
- Halbert, C.L.; Alexander, I.E.; Wolgamot, G.M.; Miller, A.D. Adeno-associated virus vectors transduce primary cells much less efficiently than immortalized cells. J. Virol. 1995, 69, 1473–1479.
- Teramoto, S.; Johnson, L.G.; Huang, W.; Leigh, M.W.; Boucher, R.C. Effect of adenoviral vector infection on cell proliferation in cultured primary human airway epithelial cells. Hum. Gene Ther. 1995, 6, 1045–1053.
- Rubenstein, R.C.; Egan, M.E.; Zeitlin, P.L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Investig. 1997, 100, 2457–2465.
- Mosler, K.; Coraux, C.; Fragaki, K.; Zahm, J.M.; Bajolet, O.; Bessaci-Kabouya, K.; Puchelle, E.; Abély, M.; Mauran, P. Feasibility of nasal epithelial brushing for the study of airway epithelial functions in CF infants. J. Cyst. Fibros. 2008, 7, 44–53.
- Scholte, B.J.; Kansen, M.; Hoogeveen, A.T.; Willemse, R.; Rhim, J.S.; van der Kamp, A.W.; Bijman, J. Immortalization of nasal polyp epithelial cells from cystic fibrosis patients. Exp. Cell Res. 1989, 182, 559–571.
- Claass, A.; Claus, S.; Höft, J.; Prange, E. Long term culture of nasal epithelial cells. Acta Histochem. 1991, 90, 21–26.
- Gruenert, D.C.; Basbaum, C.B.; Widdicombe, J.H. Long-term culture of normal and cystic fibrosis epithelial cells grown under serum-free conditions. In Vitro Cell. Dev. Biol. 1990, 26, 411–418.
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607.
- Reynolds, S.D.; Rios, C.; Wesolowska-Andersen, A.; Zhuang, Y.; Pinter, M.; Happoldt, C.; Hill, C.L.; Lallier, S.W.; Cosgrove, G.P.; Solomon, G.M.; et al. Airway Progenitor Clone Formation Is Enhanced by Y-27632-Dependent Changes in the Transcriptome. Am. J. Respir. Cell Mol. Biol. 2016, 55, 323–336.
- Kaneko, Y.; Konno, T.; Kohno, T.; Kakuki, T.; Miyata, R.; Ohkuni, T.; Kakiuchi, A.; Yajima, R.; Ohwada, K.; Kurose, M.; et al. Induction of airway progenitor cells via p63 and KLF11 by Rho-kinase inhibitor Y27632 in hTERT-human nasal epithelial cells. Am. J. Transl. Res. 2019, 11, 599–611.
- Wolf, S.; Perez, G.F.; Mukharesh, L.; Isaza, N.; Preciado, D.; Freishtat, R.J.; Pillai, D.; Rose, M.C.; Nino, G. Conditional reprogramming of pediatric airway epithelial cells: A new human model to investigate early-life respiratory disorders. Pediatr. Allergy Immunol. 2017, 28, 810–817.
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J., 3rd; Ostmann, A.J.; Strecker, L.M.; Szczesniak, R.D.; Clancy, J.P. Detection of CFTR function and modulation in primary human nasal cell spheroids. J. Cyst. Fibros. 2018, 17, 26–33.
- Awatade, N.T.; Wong, S.L.; Capraro, A.; Pandzic, E.; Slapetova, I.; Zhong, L.; Turgutoglu, N.; Fawcett, L.K.; Whan, R.M.; Jaffe, A.; et al. Significant functional differences in differentiated Conditionally Reprogrammed (CRC)- and Feeder-free Dual SMAD inhibited-expanded human nasal epithelial cells. J. Cyst. Fibros. 2021.
- Wiszniewski, L.; Jornot, L.; Dudez, T.; Pagano, A.; Rochat, T.; Lacroix, J.S.; Suter, S.; Chanson, M. Long-term cultures of polarized airway epithelial cells from patients with cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 34, 39–48.
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005, 107, 183–206.
- Whitcutt, M.J.; Adler, K.B.; Wu, R. A biphasic chamber system for maintaining polarity of differentiation of cultured respiratory tract epithelial cells. In Vitro Cell. Dev. Biol. 1988, 24, 420–428.
- Cobb, B.R.; Ruiz, F.; King, C.M.; Fortenberry, J.; Greer, H.; Kovacs, T.; Sorscher, E.J.; Clancy, J.P. A(2) adenosine receptors regulate CFTR through PKA and PLA(2). Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L12–L25.
- Sun, H.; Harris, W.T.; Kortyka, S.; Kotha, K.; Ostmann, A.J.; Rezayat, A.; Sridharan, A.; Sanders, Y.; Naren, A.P.; Clancy, J.P. Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS ONE 2014, 9, e106842.
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620.
- Bratcher, P.E.; Yadav, S.; Shaughnessy, C.A.; Thornell, I.M.; Zeitlin, P.L. Effect of apical chloride concentration on the measurement of responses to CFTR modulation in airway epithelia cultured from nasal brushings. Physiol. Rep. 2020, 8, e14603.
- Schögler, A.; Blank, F.; Brügger, M.; Beyeler, S.; Tschanz, S.A.; Regamey, N.; Casaulta, C.; Geiser, T.; Alves, M.P. Characterization of pediatric cystic fibrosis airway epithelial cell cultures at the air-liquid interface obtained by non-invasive nasal cytology brush sampling. Respir. Res. 2017, 18, 215.
- Coles, J.L.; Thompson, J.; Horton, K.L.; Hirst, R.A.; Griffin, P.; Williams, G.M.; Goggin, P.; Doherty, R.; Lackie, P.M.; Harris, A.; et al. A Revised Protocol for Culture of Airway Epithelial Cells as a Diagnostic Tool for Primary Ciliary Dyskinesia. J. Clin. Med. 2020, 9, 3753.
- Wu, Y.S.; Jiang, J.; Ahmadi, S.; Lew, A.; Laselva, O.; Xia, S.; Bartlett, C.; Ip, W.; Wellhauser, L.; Ouyang, H.; et al. ORKAMBI-Mediated Rescue of Mucociliary Clearance in Cystic Fibrosis Primary Respiratory Cultures Is Enhanced by Arginine Uptake, Arginase Inhibition, and Promotion of Nitric Oxide Signaling to the Cystic Fibrosis Transmembrane Conductance Regulator Channel. Mol. Pharmacol. 2019, 96, 515–525.
- Gianotti, A.; Capurro, V.; Scudieri, P.; Galietta, L.J.; Moran, O.; Zegarra-Moran, O. Pharmacological rescue of mutant CFTR protein improves the viscoelastic properties of CF mucus. J. Cyst. Fibros. 2016, 15, 295–301.
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375.
- McCarthy, C.; Brewington, J.J.; Harkness, B.; Clancy, J.P.; Trapnell, B.C. Personalised CFTR pharmacotherapeutic response testing and therapy of cystic fibrosis. Eur. Respir. J. 2018, 51.
- Park, J.K.; Shrivastava, A.; Zhang, C.; Pollok, B.A.; Finkbeiner, W.E.; Gibb, E.R.; Ly, N.P.; Illek, B. Functional Profiling of CFTR-Directed Therapeutics Using Pediatric Patient-Derived Nasal Epithelial Cell Models. Front. Pediatr. 2020, 8, 536.
- de Courcey, F.; Zholos, A.V.; Atherton-Watson, H.; Williams, M.T.; Canning, P.; Danahay, H.L.; Elborn, J.S.; Ennis, M. Development of primary human nasal epithelial cell cultures for the study of cystic fibrosis pathophysiology. Am. J. Physiol. Cell Physiol. 2012, 303, C1173–C1179.
- Debley, J.S.; Barrow, K.A.; Rich, L.M.; Singh, P.; McKone, E.F.; Nichols, D.P. Correlation between Ivacaftor-induced CFTR Activation in Airway Epithelial Cells and Improved Lung Function: A Proof-of-Concept Study. Ann. Am. Thorac. Soc. 2020, 17, 1024–1027.
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Olshansky, S.; Moreno, C.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479.
- Ahmadi, S.; Bozoky, Z.; Di Paola, M.; Xia, S.; Li, C.; Wong, A.P.; Wellhauser, L.; Molinski, S.V.; Ip, W.; Ouyang, H.; et al. Phenotypic profiling of CFTR modulators in patient-derived respiratory epithelia. NPJ Genom. Med. 2017, 2, 12.
- Pedersen, P.S.; Procida, K.; Larsen, P.L.; Holstein-Rathlou, N.H.; Frederiksen, O. Water permeability in human airway epithelium. Pflug. Arch. 2005, 451, 464–473.
- Guimbellot, J.S.; Leach, J.M.; Chaudhry, I.G.; Quinney, N.L.; Boyles, S.E.; Chua, M.; Aban, I.; Jaspers, I.; Gentzsch, M. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017, 2.
- Bridges, M.A.; Walker, D.C.; Harris, R.A.; Wilson, B.R.; Davidson, A.G. Cultured human nasal epithelial multicellular spheroids: Polar cyst-like model tissues. Biochem. Cell Biol. 1991, 69, 102–108.
- Marthin, J.K.; Stevens, E.M.; Larsen, L.A.; Christensen, S.T.; Nielsen, K.G. Patient-specific three-dimensional explant spheroids derived from human nasal airway epithelium: A simple methodological approach for ex vivo studies of primary ciliary dyskinesia. Cilia 2017, 6, 3.
- Neugebauer, P.; Endepols, H.; Mickenhagen, A.; Walger, M. Ciliogenesis in submersion and suspension cultures of human nasal epithelial cells. Eur. Arch. Otorhinolaryngol. 2003, 260, 325–330.
- Gamarra, F.; Bergner, A.; Stauss, E.; Stocker, I.; Grundler, S.; Huber, R.M. Rotation frequency of human bronchial and nasal epithelial spheroids as an indicator of mucociliary function. Respiration 2006, 73, 664–672.
- Pedersen, P.S.; Frederiksen, O.; Holstein-Rathlou, N.H.; Larsen, P.L.; Qvortrup, K. Ion transport in epithelial spheroids derived from human airway cells. Am. J. Physiol. 1999, 276, L75–L80.
- Liu, Z.; Anderson, J.D.; Deng, L.; Mackay, S.; Bailey, J.; Kersh, L.; Rowe, S.M.; Guimbellot, J.S. Human Nasal Epithelial Organoids for Therapeutic Development in Cystic Fibrosis. Genes 2020, 11, 603.
- McDougall, C.M.; Blaylock, M.G.; Douglas, J.G.; Brooker, R.J.; Helms, P.J.; Walsh, G.M. Nasal epithelial cells as surrogates for bronchial epithelial cells in airway inflammation studies. Am. J. Respir. Cell Mol. Biol. 2008, 39, 560–568.
- McLellan, K.; Shields, M.; Power, U.; Turner, S. Primary airway epithelial cell culture and asthma in children-lessons learnt and yet to come. Pediatr. Pulmonol. 2015, 50, 1393–1405.
- Scudieri, P.; Musante, I.; Venturini, A.; Guidone, D.; Genovese, M.; Cresta, F.; Caci, E.; Palleschi, A.; Poeta, M.; Santamaria, F.; et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020, 9, 2090.
- Thavagnanam, S.; Parker, J.C.; McBrien, M.E.; Skibinski, G.; Shields, M.D.; Heaney, L.G. Nasal epithelial cells can act as a physiological surrogate for paediatric asthma studies. PLoS ONE 2014, 9, e85802.
- Liu, X.; Wu, Y.; Rong, L. Conditionally Reprogrammed Human Normal Airway Epithelial Cells at ALI: A Physiological Model for Emerging Viruses. Virol. Sin. 2020, 35, 280–289.
- Roberts, N.; Al Mubarak, R.; Francisco, D.; Kraft, M.; Chu, H.W. Comparison of paired human nasal and bronchial airway epithelial cell responses to rhinovirus infection and IL-13 treatment. Clin. Transl. Med. 2018, 7, 13.
- Boudewijn, I.M.; Faiz, A.; Steiling, K.; van der Wiel, E.; Telenga, E.D.; Hoonhorst, S.J.M.; Ten Hacken, N.H.T.; Brandsma, C.A.; Kerstjens, H.A.M.; Timens, W.; et al. Nasal gene expression differentiates COPD from controls and overlaps bronchial gene expression. Respir. Res. 2017, 18, 213.
- Comer, D.M.; Elborn, J.S.; Ennis, M. Comparison of nasal and bronchial epithelial cells obtained from patients with COPD. PLoS ONE 2012, 7, e32924.

