 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bridget Bax | + 3688 word(s) | 3688 | 2019-07-23 12:49:58 | | | |
| 2 | Bruce Ren | -19 word(s) | 3669 | 2020-10-28 09:00:13 | | | | |
| 3 | Bruce Ren | Meta information modification | 3669 | 2020-10-28 09:15:36 | | |
Video Upload Options
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal recessive disorder caused by mutations in the nuclear TYMP gene, which encodes for thymidine phosphorylase, an enzyme required for the normal metabolism of deoxynucleosides, thymidine, and deoxyuridine. The subsequent elevated systemic concentrations of deoxynucleosides lead to increased intracellular concentrations of their corresponding triphosphates, and ultimately mitochondrial failure due to progressive accumulation of mitochondrial DNA (mtDNA) defects and mtDNA depletion. Currently, there are no treatments for MNGIE where effectiveness has been evidenced in clinical trials. A Phase 2, multi-centre, multiple dose, open label trial without a control will investigate the application of erythrocyte-encapsulated thymidine phosphorylase (EE-TP) as an enzyme replacement therapy for MNGIE. Three EE-TP dose levels are planned with patients receiving the dose level that achieves metabolic correction. The study duration is 31 months, comprising 28 days of screening, 90 days of run-in, 24 months of treatment and 90 days of post-dose follow-up. The primary objectives are to determine the safety, tolerability, pharmacodynamics, and efficacy of multiple doses of EE-TP. The secondary objectives are to assess EE-TP immunogenicity after multiple dose administrations and changes in clinical assessments, and the pharmacodynamics effect of EE-TP on clinical assessments.
1. Introduction
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a fatal and rare autosomal recessive disorder of nucleotide metabolism caused by mutations in the nuclear thymidine phosphorylase gene (TYMP), which encodes cytosolic thymidine phosphorylase, the enzyme required for the normal metabolism of pyrimidine deoxynucleosides, thymidine, and deoxyuridine [1][2]. Pathogenic variants in TYMP result in a complete or partial absence of thymidine phosphorylase activity (<10% of healthy unaffected individuals), leading to an accumulation of thymidine and deoxyuridine in tissues and body fluids[3][4][5][6][7][8][9].
Elevated systemic concentrations of these deoxynucleosides lead to increased intracellular concentrations of their corresponding triphosphates. This perturbs the physiological equilibrium of the deoxynucleoside triphosphate pools within the mitochondria, thereby interfering with the normal replication of mitochondrial mtDNA, leading to multiple deletions, somatic point mutations and depletion of mtDNA [5][8][10][11], and ultimately mitochondrial failure [5][6][8]. It is believed that the consequent failure of cellular energy production directly causes the cardinal clinical manifestations through damage to the nervous and muscular systems.
Patients with MNGIE usually present during the second decade of life, although patients have presented as early as five months and as late as the fifth decade; the average age at diagnosis is 18.5 years[12] . The relatively late onset for a condition present at birth is thought to be due to the progressive accumulation of mtDNA defects, with the disease becoming apparent once the number of affected mitochondria reaches a critical threshold level. The disease is a multi-system disorder, and has a characteristic, although by no means universal, clinical presentation. Patients typically present with gastrointestinal symptoms including early satiety, nausea, dysphagia, gastroesophageal reflux, postprandial emesis, episodic abdominal pain, episodic abdominal distention, and diarrhoea. These symptoms are secondary to alimentary dysmotility caused by degeneration of the alimentary autonomic nervous system [2]. Patients generally have a thin body habitus with reduced muscle mass and cachexia. Episodes of frank intestinal pseudo obstruction may occur, and some patients develop a hepatopathy with liver steatosis and cirrhosis. Progressive external ophthalmoplegia and peripheral sensorimotor polyneuropathy are invariable. The latter affects the lower limbs initially and is typically demyelinating. On magnetic resonance imaging (MRI) there is, in the majority of cases, leukoencephalopathy with diffuse increased T2 signal in the deep white matter of the cerebral hemispheres, but this is generally believed to be asymptomatic[5][12].
MNGIE is a progressive disease, with patients dying at an average age of 37.5 years, and at present there are no approved therapies [4]. Allogeneic hematopoietic stem cell transplantation (HSCT) offers the possibility of a permanent correction of the thymidine phosphorylase deficiency. However, it is still highly experimental, carrying a mortality rate of approximately 63%[13] . Treatment with allogeneic HSCT is limited by the availability of a matched donor, and patients are often in poor clinical condition with impaired capacity to tolerate transplant related problems and the aggressive conditioning and immunosuppressive chemotherapy[13][14]. The administration of HSCT to patients with MNGIE presents pharmacological challenges in terms of administering drugs with possible mitochondrial toxicity, and the requirement for parenteral administration due to disturbed gastrointestinal function and impairment of absorption. A published consensus proposal for standardising an approach to allogeneic HSCT in patients with MNGIE recommends restricting the recruitment of patients with an optimal donor to those without irreversible end stage disease[13][14]. Patients who are oligosymptomatic are often reluctant to undergo HSCT due to its high mortality risk. Many patients are therefore ineligible for this treatment option and clinical management is based on symptom relief and palliation. A second experimental permanent treatment approach for MNGIE is orthotopic liver transplantation. Sustained normalisation of plasma thymidine and deoxyuridine concentrations have been reported in two patients who received liver transplantation [15]. A longitudinal evaluation of additional transplanted patients, however, is essential to confirm the clinical efficacy of this treatment approach. There is thus a critical requirement to develop an alternative treatment for these patients which would provide an expeditious normalization of nucleosides to prevent as much mitochondrial damage as possible.
The Investigational Medicinal Product under investigation in this study is erythrocyte encapsulated thymidine phosphorylase (EE-TP), which is produced under Good Manufacturing Practice by the ex vivo encapsulation of recombinant Escherichia coli thymidine phosphorylase into patient’s autologous erythrocytes using an automated red cell loader device [16]. EE-TP is intravenously infused into the patient where it aims to correct the fundamental lesion in MNGIE by replacement of the deficient thymidine phosphorylase. The rationale for the development of EE-TP is based on thymidine and deoxyuridine being able to diffuse across the erythrocyte membrane via nucleoside transporters into the cytosol where the encapsulated enzyme catalyses their metabolism to the normal products, thymine and uracil, respectively (Figure 1). The products then exit the cell into the blood plasma where they are further metabolised as normal. It is proposed that regular intravenous (IV) administrations of EE-TP to patients with MNGIE will lead to a sustained reduction or elimination of plasma thymidine and deoxyuridine, leading to a clearance from the cellular compartments and thus an amelioration of the intracellular deoxynucleotide imbalances. This should prevent further damage to mtDNA. By relieving the nervous system and muscle of the toxic effects of the accumulated metabolites, EE-TP aims to arrest and reverse the progression of the clinical disease.
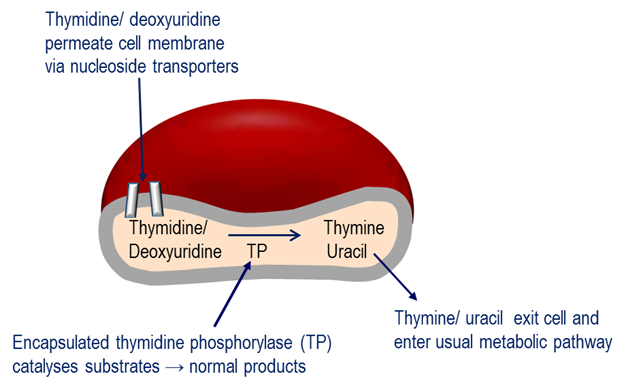
Figure 1. Mechanism of erythrocyte encapsulated thymidine phosphorylase (EE-TP) action. Plasma thymidine and deoxyuridine diffuse across the erythrocyte membrane via nucleoside transporters into the erythrocyte cytosol where the encapsulated thymidine phosphorylase (TP) catalyses their metabolism to thymine and uracil, which then exit the erythrocyte to enter the normal metabolic pathways.
EE-TP has the advantage of prolonging the circulatory half-life of the enzyme to that of the erythrocyte half-life (19 to 29 days) and minimising immunogenic reactions, which are often observed in enzyme replacement therapies administered by the conventional route.
Clinical experience with EE-TP is limited to a proof-of-concept study in a single patient diagnosed with MNGIE and a compassionate clinical evaluation in 4 patients with MNGIE [17][18][19]. EE-TP was well tolerated and reductions in the disease-associated plasma metabolites, thymidine and deoxyuridine, were observed in all patients. Clinical improvements were observed in three patients who received long-term treatment, suggesting that EE-TP is able to reverse some aspects of the disease pathology. Transient, non-serious adverse events were observed in two of the five patients; these did not lead to therapy discontinuation and were managed with pre-medication prior to infusion of EE-TP.
The aim of this study is to investigate the safety, tolerability, pharmacodynamics, and efficacy of EE-TP in patients with MNGIE. We hypothesise that treatment with EE-TP will arrest and reverse the progression of the clinical disease. The study protocol reported here (version 6.0) was written in compliance with the Standard Protocol Items: recommendations for Interventional Trials (SPIRIT) 2013 [20]. The sponsor, St George’s, University of London requested scientific assistance for EE-TP and the current protocol was updated in line with the European Medicines Agency (EMA) advice provided. The study will be conducted in accordance with the following and all subsequent amendments: i) ICH E6: Good Clinical Practice: Consolidated guideline Committee for Proprietary Medicinal Products (CPMP)/ICH/135/95 (July 1996), adopted in the EU by CPMP, ii) European Commission Directive 2001/20/EC (April 2001), iii) European Commission Directive 2003/94/EC (October 2003), iv) European Commission Directive 2005/28/EC (April 2005), v) Manufacture of Investigational Medicinal Products: Volume 4, Annex 13 of the EU Guidelines to GMP (February 2010) and vi) The Medicines for Human Use (Clinical Trials) Regulations 2004 and all subsequent amendments.
Study Synopsis
|
Title of Study: |
A Multi‑centre, Multiple‑dose, Open‑label Study to Investigate the Safety, Tolerability, Pharmacodynamics, and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase (EE‑TP) in Patients with Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) |
|
Indication: |
Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) |
|
Study Centres: |
The study will be conducted at sites in Europe and Israel. |
|
Development Phase: |
II |
|
Objectives: |
Primary Objective: The primary objective is to determine the safety, tolerability, pharmacodynamics (PD), and efficacy (as measured by weight stabilisation) of multiple doses of EE‑TP manufactured using the red cell loader in patients with MNGIE. Secondary Objectives: The secondary objectives are: To assess the immunogenicity of EE‑TP after multiple‑dose administrations. To assess changes in clinical assessments. To assess the PD effect of EE‑TP on clinical assessments. Exploratory Objective: The exploratory objective is to assess the effects of EE‑TP on markers of mitochondrial condition, In addition, MRI (brain), abdominal ultrasound, electromyography, and neuro-ophthalmological assessments are exploratory too. |
|
Study Population: |
Patients with MNGIE |
|
Methodology/ |
This will be a multi‑centre, multiple‑dose, open‑label study to investigate the safety, tolerability, PD, and efficacy of EE‑TP in patients with MNGIE. The study will be conducted in an open‑label manner with all patients receiving EE‑TP. A Patient Oversight Committee (POC) will perform ongoing reviews of safety, tolerability, efficacy and available PD data to ensure the acceptability of continued dosing, and/or dose progression for each patient. Additional ad hoc POC meetings may take place to discuss time‑sensitive issues, as required. An Independent Data Monitoring Committee (IDMC) will perform ongoing reviews of safety, tolerability, efficacy and available PD data at predefined decision points. The IDMC will also provide recommendations on the need for additional patients, progression to younger age groups, modifications to the study and dosing after treatment cycle six in the event of no metabolic correction. A steering committee comprising representatives of the Sponsor and Chief Investigator will review reports from the IDMC and POC. |
|
Number of Patients (Planned and Analysed): |
The study will enrol 12 adult treatment‑naïve patients with MNGIE, aged 18 years or older at Screening. With IDMC approval, a further four juvenile patients (aged 16-17) will be recruited after at least 24 patient-months exposure to treatment. With IDMC approval, a further four juvenile patients (aged 12-15) will be recruited after at least 24 patient-months exposure to treatment in the 16-17 year old patient group. Screening failures and patients withdrawing from the study may be substituted with IDMC approval. Patients who have previously received EE‑TP on a ‘compassionate use’ basis may be included in the study, if they meet eligibility criteria; these patients will be additional and will not be included in the sample size of 12. The total sample size of 12 adult treatment‑naïve patients is not based on a formal statistical assessment, but is dictated by practical considerations mainly due to the rarity of the condition. |
|
Diagnosis and Main Criteria for Inclusion: |
Patients must be aged 18 years or older at Screening and may be male or female of any race. Following IDMC review of the benefit‑risk profile, the age range will be extended to include (i) patients aged 16 years or older after at least 24 patient‑months of exposure in those aged 18 years or over, and (ii) patients aged 12 years or older after at least 24 patient‑months of exposure in patients of <18 years of age at the time of enrolment. Patients must be diagnosed with MNGIE by demonstrating ALL of the following: <18% normal thymidine phosphorylase (TP) activity in the buffy coat; >3 μmol/L plasma thymidine; >5 μmol/L plasma deoxyuridine; · Confirmation of the presence of a pathogenic mutation in the TP gene by sequencing. |
|
Exclusion Criteria: |
Patients who meet any of the following criteria will not be eligible to participate in the study: 1. Patients who have received a successful liver or bone marrow transplant. 2. Patients suitable for AHSCT. 3. Patients with a matched AHSCT donor. 4. Patients with a known history of human immunodeficiency virus, hepatitis B infection, or an active hepatitis C infection. 5. Patients who are severely disabled (e.g., patient bed‑bound, incontinent, and unable to carry out any daily activities), or with a life expectancy of less than 12 months at Screening, based on the Investigator’s judgment. 6. Female patients who are: a. pregnant, planning a pregnancy, or are unwilling to use contraception b. Breastfeeding or lactating. 7. Patients who have donated blood in the 90 days prior to Screening. 8. Patients with a confirmed red blood cell (RBC) count of <3.0 × 109 per mL. 9. Patients who have a significant history of alcoholism or drug/chemical abuse within 1 year prior to Screening, as determined by the Investigator. 10. Patients who have an abnormality in heart rate, blood pressure, or body temperature at Screening that, in the opinion of the Investigator, increases the risk of participating in the study. 11. Patients who have an abnormality in the 12‑lead electrocardiogram (ECG) at Screening that, in the opinion of the Investigator, increases the risk of participating in the study. 12. Patients who have, or have a history of, any clinically significant neurological, gastrointestinal (GI), renal, hepatic, cardiovascular, psychiatric, respiratory, metabolic, endocrine, haematological, or other major disorder (except for MNGIE, or disorders associated with MNGIE that, in the Investigator’s opinion, do not constitute a risk when taking study drug and would not interfere with the study objectives) as determined by the Investigator. 13. Patients with any current malignancy, or a history of malignancy within 5 years prior to Screening, with the exception of adequately treated or excised non‑metastatic basal cell or squamous cell cancer of the skin or cervical carcinoma in situ. 14. Patients who are currently enrolled in, or are planning to participate in, or discontinued within the last 30 days from a clinical study involving an investigational medicinal product or concurrently enrolled in medical research judged not to be scientifically or medically compatible with EE‑TP. 15. Patients with any medical condition, which in the opinion of the Investigator, would make the patient unsuitable for enrolment or could interfere with the patient’s participation in, or completion of, the study. |
|
Study Medications: |
All patients will be administered EE‑TP by intravenous (IV) infusion. There are 3 planned dose levels: Dose Level 1 (low dose): ~58 -65 x1010 erythrocytes encapsulating 30 to 49 U TP/1010 erythrocytes Dose Level 2 (mid dose): ~58 -65 x1010 erythrocytes encapsulating 50 to 69 U TP/1010 erythrocytes Dose Level 3 (high dose): ~58 -65 x1010 erythrocytes encapsulating 70 to 90 U TP/1010 erythrocytes All patients will receive infusions at Dose Level 1 (low dose) for the first 2 treatment cycles. If metabolic correction is not achieved, the subsequent 2 treatment cycles will be infusions at Dose Level 2 (mid dose). If metabolic correction is still not achieved, treatment will advance to Dose Level 3 (high dose) for subsequent treatment cycles. If, at any time during this progression, metabolic correction is achieved, the patient will continue at the Dose Level in which metabolic correction has been achieved and there will be no further dose escalation. Patients will be administered EE‑TP every 3 weeks until the dose level achieving metabolic correction is identified. From Day 78 (or once metabolic correction has been achieved), it is planned that patients will receive EE‑TP every 2 to 4 weeks until the end of the study. The interval between doses will not be shorter than 2 weeks ±2 days. Dose selection will be flexible; continued PD assessment will enable dose optimisation and further inform dose‑response models and establish the therapeutic window for the treatment. |
|
Dose and Regimen: |
Single doses of EE‑TP will be administered on Day 0 then every 3 weeks until metabolic correction is achieved. The starting dose will be Dose Level 1, with potential subsequent dose levels of Dose Level 2 and Dose Level 3 dependent on whether metabolic correction is achieved or not. All doses will be administered IV. From Day 78 (or once metabolic correction has been achieved), it is planned that patients will receive EE‑TP every 2 to 4 weeks until the end of the study. Dose frequency may be reduced (e.g., from every 2 weeks to every 3‑4 weeks) for individual patients based on ongoing review of emerging safety, tolerability, efficacy, and PD data. The interval between doses will not be shorter than 2 weeks ±2 days. The dose level from day 78 onwards will remain at the dose that achieved metabolic correction during the initial treatment phase (pre-day 78). In the advent of an increase in plasma metabolite levels (i.e. loss of metabolic correction), the frequency of dosing and/or dose level will be reviewed and adjusted accordingly. |
|
Duration of Treatment: |
Infusions with EE‑TP will occur once every 2 to 4 weeks for 24 months. |
|
Duration of Patient Participation in Study: |
Planned Enrolment/Screening (Run‑in) Duration: approximately 28 days (Days ‑120 to Day ‑92). Run-in period of 90 days (Days -91 to 0) for repeated evaluations of a number of assessments. Follow up, 90 days post dose. |
|
Study Populations: |
The All Patients Population will consist of any patients who enrolled on to the study (signed informed consent) and had study assessments recorded on the database as per the protocol. The Safety Population will consist of all patients who received at least 1 dose of EE‑TP and have at least 1 post dose safety assessment. The Efficacy Population will consist of all patients who received at least 1 dose of EE‑TP and have evaluable efficacy data. The Pharmacodynamic Population will consist of all patients who received at least 1 dose of EE‑TP and have evaluable PD data. All protocol deviations will be assessed for severity/impact. The decisions leading to exclusion of patients from population analyses will be based on these assessments, made prior to database lock and documented in the clinical study report. |
|
Evaluation: |
The safety of EE‑TP will be evaluated by: incidence, frequency, and severity of adverse events (or treatment‑emergent adverse events [TEAEs]) incidence of laboratory abnormalities, based on haematology, serum biochemistry, and urinalysis test results vital sign measurements 12‑lead ECG parameters physical examination findings use of concomitant medication(s) |
|
Evaluation: |
The following biomarkers will be evaluated for assessment of the PD effect(s) of EE‑TP: Urine and plasma thymidine concentrations Urine and plasma deoxyuridine concentrations Anti-thymidine phosphorylase antibodies |
|
Evaluation: |
The efficacy of EE‑TP will be assessed by the following: Primary efficacy endpoint: Mean absolute change from baseline in body mass index (BMI) at 24 months Secondary efficacy endpoints: Mean absolute change from baseline BMI at day 63 and 3, 6, 9, 12, 15, 18, and 21 months Change from baseline in the proportion of patients who require total parenteral nutrition or have deceased at day 63 and 3, 6, 9, 12, 15, 18, 21, and 24 months Mean absolute change from baseline handgrip strength measured using dynamometer (for assessment of distal muscle weakness) employed according to the Southampton protocol for adult grip strength at 6, 12, 18, and 24 months (for both adults and juvenile patients) Mean absolute change from baseline disability measured using the Rasch‑built Overall Disability Scale at 6, 12, 18, and 24 months Mean absolute change from baseline ambulatory function measured using the timed 10‑metre walk test at 6, 12, 18, and 24 months Mean absolute change from baseline quality of life measured using EuroQol‑5 Dimension and Clinical Global Impression – Improvement Scale at 6, 12, 18, and 24 months Mean absolute change from baseline of GI symptoms measured using PROMIS® short form scales (GI belly pain, GI diarrhoea, GI disrupted swallowing, GI gas and bloating, GI gastroesophageal reflux, and GI nausea and vomiting) at 6, 12, 18, and 24 months Patient Global Impression of Change at 6, 12, 18, and 24 months, relative to baseline Changes relative to baseline in motor strength, distal sensory impairment, or areflexia using neurological exam tests recorded in a standardised fashion Improvement of the most disabling symptom for each patient (assessed using the visual analogue scale) |
|
Statistical Methods: |
Adverse event summaries will include tabulations of all TEAEs, TEAEs by maximum severity, and TEAEs by relationship to study drug. By patient listings will be provided for deaths, serious adverse events, DLTs, and adverse events leading to discontinuation from study treatment. Laboratory parameter (haematology, serum biochemistry, and urinalysis) shifts from baseline to low or high post‑baseline values will be presented. Mean laboratory and vital signs values over time will be plotted by dose level. Descriptive statistics will be provided for clinical laboratory data and vital signs. Electrocardiogram data, physical examination data and use of concomitant medication will be listed by patient. Pharmacodynamic data, including urine and plasma thymidine and deoxyuridine will be assessed descriptively whenever possible, and summary statistics will be provided. Associations between PD parameters and clinical activity parameters will be explored. Absolute and changes from baseline in each efficacy endpoint will be listed. Absolute and changes from baseline in BMI will be plotted against plasma and urine concentrations of thymidine and deoxyuridine. The proportion of patients achieving at least 0.5 kg will be presented by dose. Interim reviews of open‑label data will be conducted after all 12 patients in the study have completed at least 1 year of EE-TP dosing. |
References
- . Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE): A Disease of Two Genomes . Neurologist. Retrieved 2019-7-23
- Hirano, M. Mitochondrial Neurogastrointestinal Encephalopathy Disease; Adam MP, Ardinger HH, Pagon RA, Eds.; University of Washington: Seattle, USA, 1993; pp. 1.
- Ichizo Nishino; Thymidine Phosphorylase Gene Mutations in MNGIE, a Human Mitochondrial Disorder. Science 1999, 283, 689-692, 10.1126/science.283.5402.689.
- Ichizo Nishino; Antonella Spinazzola; Alexandros Papadimitriou; Simon Hammans; Israel Steiner; Cecil D. Hahn; Anne M. Connolly; Alain Verloes; João Guimarães; Ivan Maillard; et al.Hitoshi HamanoM. Alice DonatiCarol E. SemradJames A. RussellAntonio L. AndreuGiorgos M. HadjigeorgiouTuan H. VuSaba TadesseTorbjoern G. NygaardIkuya NonakaIkuo HiranoEduardo BonillaLewis P. RowlandSalvatore DiMauroMichio Hirano Mitochondrial neurogastrointestinal encephalomyopathy: An autosomal recessive disorder due to thymidine phosphorylase mutations. Annals of Neurology 2000, 47, 792-800, 10.1002/1531-8249(200006)47:6<792::aid-ana12>3.3.co;2-p.
- M. Hirano; Gabriella Silvestri; D. M. Blake; A. Lombès; C. Minetti; E. Bonilla; A. P. Hays; R. E. Lovelace; I. Butler; T. E. Bertorini; et al.A. B. ThrelkeldH. MitsumotoL. M. SalbergL. P. RowlandS. DiMauro Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 1994, 44, 721-721, 10.1212/wnl.44.4.721.
- A. Spinazzola; Ramon Marti; Ichizo Nishino; A. L. Andreu; A. Naini; S. Tadesse; I. Pela; E. Zammarchi; M. A. Donati; J. A. Oliver; et al.M. Hirano Altered Thymidine Metabolism Due to Defects of Thymidine Phosphorylase. Journal of Biological Chemistry 2001, 277, 4128-4133, 10.1074/jbc.m111028200.
- Ramon Marti; Definitive Diagnosis of Mitochondrial Neurogastrointestinal Encephalomyopathy by Biochemical Assays. Clinical Chemistry 2004, 50, 120-124, 10.1373/clinchem.2003.026179.
- Ramon Martı́; Yutaka Nishigaki; Michio Hirano; Elevated plasma deoxyuridine in patients with thymidine phosphorylase deficiency. Biochemical and Biophysical Research Communications 2003, 303, 14-18, 10.1016/s0006-291x(03)00294-8.
- Maria Lucia Valentino; Ramon Martí; Saba Tadesse; Luis Carlos López; Jose L. Manes; Judy Lyzak; Angelika Hahn; Valerio Carelli; Michio Hirano; Thymidine and deoxyuridine accumulate in tissues of patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). FEBS Letters 2007, 581, 3410-3414, 10.1016/j.febslet.2007.06.042.
- Yutaka Nishigaki; Ramón Martí; William C. Copeland; Michio Hirano; Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. Journal of Clinical Investigation 2003, 111, 1913-1921, 10.1172/JCI200317828.
- Y. Nishigaki; Ramon Martí; Michio Hirano; ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Human Molecular Genetics 2003, 13, 91-101, 10.1093/hmg/ddh010.
- Ichizo Nishino; Antonella Spinazzola; Michio Hirano; MNGIE: from nuclear DNA to mitochondrial DNA. Neuromuscular Disorders 2001, 11, 7-10, 10.1016/s0960-8966(00)00159-0.
- Joerg P. Halter; W. Michael; M. Schüpbach; Hanna Mandel; Carlo Casali; Kim Orchard; Matthew Collin; David Valcarcel; Attilio Rovelli; Massimiliano Filosto; et al.Maria T. DottiGiuseppe MarottaGuillem PintosPere BarbaAnna AccarinoChristelle FerraIsabel IllaYves BeguinJaap A. BakkerJaap Jan BoelensIrenaeus F. M. De CooKeith FayCarolyn M. SueDavid NachbaurHeinz ZollerCláudia SobreiraBelinda Pinto SimoesSimon R. HammansDavid SavageRamon MartíPatrick F. ChinneryRonit ElhasidAlois GratwohlMichio HiranoGuillem Pintos-Morell Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 2015, 138, 2847-2858, 10.1093/brain/awv226.
- Halter, J.; Schüpbach, W. M. M.; Casali, C.; Elhasid, R.; Fay, K.; Hammans, S.; Illa, I.; Kappeler, L.; Krähenbühl, S.; Lehmann, T; et al. Allogeneic Hematopoietic SCT as Treatment Option for Patients with Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE): A Consensus Conference Proposal for a Standardized Approach. . Bone Marrow Transplant 2011, 46, 330-337, https://doi.org/10.1038/bmt.2010.100.
- Roberto D'angelo; Rita Rinaldi; Loris Pironi; Maria Teresa Dotti; Antonio Daniele Pinna; Elisa Boschetti; Mariantonietta Capristo; Susan Mohamed; Manuela Contin; Leonardo Caporali; et al.Valerio CarelliRoberto De Giorgio Liver transplant reverses biochemical imbalance in mitochondrial neurogastrointestinal encephalomyopathy. Mitochondrion 2017, 34, 101-102, 10.1016/j.mito.2017.02.006.
- M Magnani; L Rossi; M D'ascenzo; I Panzani; L Bigi; A Zanella; Erythrocyte engineering for drug delivery and targeting.. Biotechnology and Applied Biochemistry 1998, 28, 1-6, https://doi.org/10.1111/j.1470-8744.1998.tb00505.x.
- N. F. Moran; M. D. Bain; Miratul Muqit; B. E. Bax; CARRIER ERYTHROCYTE ENTRAPPED THYMIDINE PHOSPHORYLASE THERAPY FOR MNGIE. Neurology 2008, 71, 686-688, 10.1212/01.wnl.0000324602.97205.ab.
- Bridget E. Bax; Murray D. Bain; Mauro Scarpelli; Massimiliano Filosto; Paola Tonin; Nicholas Moran; Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology 2013, 81, 1269-1271, 10.1212/WNL.0b013e3182a6cb4b.
- Michelle Levene; Murray D. Bain; Nicholas F. Moran; Niranjanan Nirmalananthan; Joanna Poulton; Mauro Scarpelli; Massimiliano Filosto; Hanna Mandel; Andrew D. MacKinnon; Lynette Fairbanks; et al.Dario PacittiBridget Bax Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in Mitochondrial Neurogastrointestinal Encephalomyopathy. Journal of Clinical Medicine 2019, 8, 457, 10.3390/jcm8040457.


