 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fatma Dogan | + 3321 word(s) | 3321 | 2021-04-21 05:50:14 | | | |
| 2 | Peter Tang | Meta information modification | 3321 | 2021-04-29 10:26:03 | | |
Video Upload Options
Maintenance of telomeres is a fundamental step in human carcinogenesis and is primarily regulated by telomerase and the human telomerase reverse transcriptase gene (TERT). Improved understanding of the transcriptional control of this gene may provide potential therapeutic targets. Epigenetic modifications are a prominent mechanism to control telomerase activity and regulation of the TERT gene.
1. Telomeres and Telomerase Regulation
Telomerase, a ribonucleoprotein enzyme, is the main complex responsible for telomere elongation. The telomeric sequence was recognized as an essential structure at the end of chromosomes [1][2], then sequenced and identified as tandem repeats in the unicellular eukaryote Tetrahymena in 1978 [3]. The telomerase enzyme was discovered in the same model organism [4], followed shortly afterward in human cells [5]. Following on from these pivotal discoveries a substantial research effort has focused on the role of telomerase and telomeres in ageing, cancer and disease. The telomerase complex consists of two main subunits and a range of associated proteins such as Ku, HSP90 and telomerase-associated protein (TP1) [6][7]. The main subunits are the highly conserved, catalytic, human telomerase reverse transcriptase (TERT) protein (1132 amino acids, 127 kDa) [8] and the telomerase RNA component (TERC), where both components are required for telomerase activity [9]. A telomerase-associated protein example, DNA repair protein Ku (a heterodimer of Ku70 and Ku80 subunits), interacts with telomerase through interaction with TERT and TERC subunits [10][11]. TERC component and telomerase-associated proteins are constitutively expressed [12] indicating that enzyme activity depends on transcriptional regulation of TERT which is the rate-limiting component of telomerase activity. A direct correlation between TERT mRNA level expression and telomerase activity is well described [13].
Genetic mechanisms contribute to the regulation of TERT in cancer through promoter mutations [14], gene copy number variations [15], and genomic rearrangements [16]. Epigenetic modifications through promoter methylation [17], histone acetylation and methylation, transcriptional and posttranscriptional mechanisms, and non-coding RNAs [18] all also contribute to the regulation of TERT expression in human tumour cell lines [19][20][21].
Telomeres are guanine-rich tandem repeats that protect chromosomes from degradation, provide stabilization, and contribute to overall chromosomal organization [22]. Human telomeres terminate with 3′ single-stranded-DNA overhang which forms a telomere loop stabilized by telomere binding proteins, such as TRF2 (Telomeric repeat-binding factors 2), that protect the telomere end and prevent it from being recognized as a site of DNA damage [23][24]. Telomeric Repeat Binding Factors 1 and 2 (TRF1, TRF2), TRF1-Interacting Nuclear protein 2 (TIN2), repressor activator protein 1 (RAP1), Protection of Telomeres 1 (POT1) and TPP1 telomere protection protein 1 (TPP1) proteins are necessary for telomere function and form the shelterin complex that protects the telomere structure from DNA damage [25]. TRF1, TRF2, and POT1 recognize TTAGGG repeats and the other three scaffold proteins (TIN2, TPP1 and RAP1). Shelterin complex proteins are constitutively expressed, potentially covering all telomeric DNA, and the absence of these proteins can lead to non-homologous end joining (NHEJ), homology-directed repair (HDR) [26], end-to-end fusions [27], genomic instability senescence, or apoptosis [28].
In human cell populations, telomere lengths range from ~5 to 15 kb and each cell division results in a telomere repeat loss of 25–200 bp due to the end replication problem [29][30] In DNA replication, the lagging strand, composed of short DNA fragments and RNA primers, provides a 3’ end for DNA polymerase-driven extension. The end-replication scenario is encountered during the 5`−3` synthesis of DNA because DNA polymerases can only add nucleotides to 3`OH groups. Therefore, the last primer of the lagging strand cannot be synthesized after the RNA primer is removed, resulting in a progressive decrease in telomeric repeats accompanied by cell replication [31]. Proliferative stem cells display regulated telomerase activity and have reduced telomere lengths when compared to germline or hESCs. Cellular senescence was first observed in normal human somatic fibroblasts [32]. Due to the absence of telomerase activity in human somatic cells, there is a progressive loss of telomere repeats, with repeated cell divisions, that ultimately induces an irreversible growth arrest called replicative senescence, or “M1”, when telomere shortening reaches a critical level. However, escape from senescence can occur following the dysregulation of cell cycle checkpoints, such as P53 (TP53), [33], ATM [34] and P16 [35], where progressive proliferation occurs until the cells reach a crisis, or “M2”, which is represented by high levels of apoptosis and genomic instability. Cancer cells can bypass crisis by reactivating telomerase, stabilizing/lengthening telomere ends, and reducing genomic instability levels. They may achieve immortalization via upregulation of telomerase and downregulation of tumour suppressor genes [36]. Telomerase can extend and maintain telomere repeats bypassing the end-replication problem. On the other hand, telomerase activity is not always related to telomere length in cancer cell lines, instead sustaining cell proliferation through telomere length stabilization [37].
Telomerase enzyme activity in somatic cells can be detected at very low levels [38]. However, it is constitutively expressed across germ and stem cells and can be highly expressed in many cancers. To avoid telomere shortening and bypass continued telomeric instability, tumour cells require telomere maintenance mechanisms. Telomerase enzyme activity remains the main mechanism for the maintenance of telomere repeats and 85–90% of cancer cells display telomerase activity [39]. Some immortal human cells maintain telomere length in the absence of telomerase activity with alternative lengthening of telomeres (ALT) mechanism [40]. ALT is based on DNA homologous recombination, and approximately 10–15% of cancers maintain their telomeres in this manner [41]. ALT positive cell lines display highly heterogeneous telomere lengths [42] and contain promyelocytic leukaemia nuclear bodies that comprise extrachromosomal telomeric DNA, proteins associated with DNA recombination and replication processes, and telomere binding proteins [43]. ALT mechanisms are repressed in hESCs but have been suggested to be activated during early development [44]. Telomere length is generally longer in stem cell populations than in somatic and cancer cell lines except for some ALT and telomerase positive cell lines [45].
The TERT locus is located on the short arm of human chromosome 5 (5p15.33), 2 Mb distal from the telomere [46]. Longer telomeres have been suggested to fold back on the TERT locus thereby negatively regulating expression via telomere position effect [20][47]. Kim and colleagues reported that young human cells with long telomeres have suppressed TERT via changes in epigenetic status. Telomere shortening alters histone marks and DNA methylation of promoter regions that regulate TERT expression. Human fibroblasts with long telomeres have significantly higher methylation levels in the TERT promoter region than cells with shorter telomeres. Further, aged cells with short telomeres show an increase in both active chromatin marks, H3K4 trimethylation (H3K4me3) and H3K9 acetylation (H3K9ac), across the TERT promoter. These results support the concept that telomere length-associated changes or telomere position effect might affect TERT transcription. Three-dimensional interactions between the TERT locus and the sub-telomeric 5p region are conserved in young somatic cells with elongated telomeres but 5p/TERT looping interactions become separated by gradual telomere shortening [20]. Telomere looping provides a testable hypothesis to explain how cells turn off enzyme activity during development when telomeres are long and, conversely, reactivation of telomerase in cancer cells that have shorter telomeres [48]. However, human embryonic stem cells (hESCs) have high telomerase activity and long telomeres suggesting that this may be an oversimplification.
2. Transcription Factors and Regulation of TERT Promoter
Transcriptional regulation of TERT can be driven through epigenetic mechanisms including DNA and histone modifications around the TSS of the promoter. The TERT promoter contains a GC-rich sequence and binding sites for many transcription factors but does not contain TATA or CAAT box transcriptional regulatory elements [49][50]. A number of transcription factors (TFs) are known to play roles in the regulation of TERT promoter including activators; SP1 (Specificity Protein 1), MYC, NF-κB (Nuclear Factor κB), AP1, STAT3 (Signal Transducer and Activator of Transcription 3), STAT5, PAX (Paired Box Proteins), ER (Estrogen Receptor), HIF1s, and repressors; MAD1, P53, WT1, (Wilms’ tumour 1 suppressor), SP3, CTCF factor and E2F1 [51]. MYC dimerized with MAX has an affinity for the two E-boxes (5′-CACGTG) in the TERT promoter [52], while the core promoter has five GC-boxes that are potential SP1 binding sites [53]. Both MYC and SP1 expression correlate with TERT transcription in various cancer cell lines [53]. The TERT/SP1 interplay displays further complexity via evidence of co-activation roles on DNMT3B expression in hepatocellular carcinoma (HCC) [54]. However, SP1 plays a dual role via repression of TERT promoter in normal human somatic cells [55]. Further, MYC antagonist, MAD1, competitively binds E-boxes and regulates repression of gene expression [56].
STAT3 has a key role in the expression of TERT in HCC [57], breast [58] and glioblastoma [59] where STAT3 expression levels correlated with TERT expression. STAT3 binding to the promoter in breast cancer stem cells [58] along with STAT5 to the distal promoter region resulted in the activation of TERT expression and telomerase activity [60][61]. NF-κB directly activated TERT promoter by binding to the proximal region, and indirectly via increased binding of MYC and SP1 [51][62]. Nuclear hormone receptor ER also played a role in TERT expression and enzyme activity through binding the oestrogen response element in the TERT promoter [63][64].
The hypoxia-inducible transcription factor family (HIF1A, HIF2A and HIF3A) are activated in response to sub-ambient oxygen levels and regulate adaptive cellular responses [65]. The TERT promoter has two HIF1 consensus sequences between −165 and +51 [66]. HIF1A upregulation induced telomerase activity accompanied by increased TERT transcription, driven through these two HIF1 binding motifs [67]. HIF1A and HIF2A significantly increased TERT promoter activity in renal cell carcinoma cell lines, while HIF2A alone inhibited TERT promoter activity in glioma cell lines [68]. Detailed schematic maps of transcription factors of the TERT promoter identify the sites of transcription factors binding, frequent mutation, and response elements that outline a complex landscape of activation, inactivation, and dual role regulators (SP1, HIF2A and EGR1) that can both activate and inactivate gene expression [51].
The P53 transcription factor, with SP1, and potentially others, suppressed TERT transcription via binding at −1877 and −1240 upstream of the transcription start site (UTSS). Moreover, P21, itself a downstream activation target of P53, has a significant role in regulating P53 dependent TERT suppression [69][70][71]. Other important transcription factors in the regulation of TERT are MYC and MAX which bind the E-boxes consensus sites (5′-CACGTG-3′) at positions −165 and +44 to regulate TERT activity [72]. Further, mutations have a significant effect on the transcription of the TERT gene. The cytosine to thymidine transitions at −124 bp and −146 bp are common mutations relative to ATG (start codon) [73][74]. These mutations are common across several cancers including melanoma, thyroid cancer, bladder cancer, glioblastoma, HCC and urothelial carcinomas [14][75]. Mutation of the TERT promoter can switch inactive marks to active chromatin marks. The wild-type allele exhibits the H3K27me3 mark associated with epigenetic silencing, while mutant TERT promoters exhibit the H3K4me2/3 mark, which is commonly associated with active chromatin, and enables recruitment of GABPA/B1 [76]. Alternative splicing provides a further level of complexity to the regulation of telomerase, especially during development. Alternative splicing is the process whereby mRNA can be utilized to direct the synthesis of different protein isoforms from a single gene by rearrangement of intron and exon elements. These isoforms can have distinct cellular functions [77][78][79]. Human TERT pre-mRNAs can be spliced into 22 isoforms during development but only the full-length transcript (16 exons) is functionally active [79]. A number of isoforms are translated into protein and many have a premature stop codon such as minus beta isoform [79]. The reverse transcriptase domain of TERT contains α, β, or α β alternative spliced isoforms [80]. Expression of splicing variants produces a biologically functional protein containing both α and β regions with a reported correlation between telomerase activity and TERT +α+β mRNA levels [81]. Alternative splice variant transcript expression (TERT –α+β, TERT +α–β and TERT –α–β) decreased telomerase activity due to interference from these non-functional alternate splice variants [81]. The –α splicing isoform is translated into a non-functional alternate splice variant without reverse transcriptase activity and its overexpression inhibited telomerase activity [82][83]. Developmentally, elevated telomerase activity correlated with full-length (16 exons) transcript expression in the fetal kidney until gestational week 15. After this time expression of non-functional alternate splice variant (TERT –α+β, TERT +α–β and TERT –α–β) expression becomes dominant and telomerase activity is reduced [81][84][85].
3. Epigenetic Control
Waddington defined epigenetics as the correlation between phenotypic and genotypic changes during mammalian embryonic development, and tissue-specific gene function, through studies on Drosophila melanogaster [86][87]. Traditional genetics describe genes and their functions while epigenetics provides a sequence-independent mechanism for influence on the gene expression process [88]. Currently, epigenetics is defined as a reversible and heritable alteration in gene expression that occurs without changes in the primary DNA sequence. Epigenetics can change chromatin and DNA structure via methylation of cytosine bases in DNA, posttranslational modifications of histone proteins, nucleosome remodeling, and non-coding RNAs (including microRNAs) [87] (Figure 1). Epigenetic modifications play a pivotal part in important cellular processes including differentiation, embryonic developmental programming, and the development of cancer. A common feature of tumours is aberrant gene regulation, therefore, epigenetic and genetic changes associated with the initiation and progression of cancer contribute to malignant transformation by working together [89]. Consequently, recent advances in the area of epigenetics contribute directly to our knowledge of cancer progression.
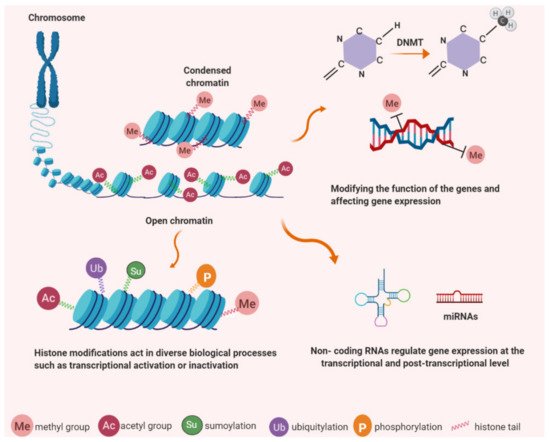
Figure 1. Epigenetic modifications have important roles in chromatin structure and gene expression. DNA is wrapped around histone proteins to form chromatin; less condensation associates with transcriptional activity while more condensed chromatin is found in a state of transcriptional silencing. Epigenetic mechanisms occur as covalent modifications of either DNA or histone proteins. DNA methylation (e.g. cytosine methylation and hydroxymethylation) and histone modification, (e.g. acetylation, methylation, phosphorylation, ubiquitination and sumoylation). Non-coding RNAs regulate protein-coding genes and associate with DNA methylation and histone modification.
Environmental factors such as pollutants, diet, temperature changes, and other external stresses can modulate the establishment of epigenetic modifications, and thereby influence subsequent gene expression, development, metabolism, and phenotype [90]. DNA methylation, for instance, has a significant role in stem cell differentiation and cellular programming. CpG methylation analysis from pluripotent cell samples (n = 269) and somatic cells demonstrated that a distinct methylation signature distinguished hiPSCs (human-induced pluripotent stem cells) and hESCs from somatic cells [91]. As highlighted above there is a range of epigenetic mechanisms including DNA methylation and packaging of DNA by histone proteins or non-coding RNAs [92]. DNA methylation describes the addition of (methylation) or oxidation (hydroxymethylation) of methyl groups, the best known epigenetic markers. These methyl groups are added to cytosine residues in DNA to form 5-methylcytosine (5mC) which can potentially block transcription factor binding access or decrease binding of gene regulatory elements resulting in reduced gene expression [93]. DNA methyltransferase (DNMT) enzymes regulate the initial process of methylation. The three major DNA methyltransferases; DNMT1, DNMT3A, and DNMT3B, drive the methylation pattern of genomic DNA [94]. DNMT1 catalyses the transfer of methyl groups to cytosine nucleotides in CpG islands of genomic DNA. The main function of DNMT1 is to maintain methylation patterns during DNA replication and it has a distinguishable preference for CpGs on hemimethylated DNA [95]. DNMT3A and DNMT3B catalyse de novo methylation of DNA sequences during gametogenesis, embryogenesis and somatic tissue development [96]. The expression level of DNMT3B is low in somatic adult cells while aberrant, elevated, DNMT3B expression is observed in several cancer cells including colorectal carcinoma, hematopoietic cell lines, bladder and breast cancer with the suggestion that DNMT3B expression is required for tumour cell survival [97][98]. DNMT3A and DNMT3B, are expressed at high levels in undifferentiated human embryonic stem cells, but subsequently down-regulated during differentiation [99][100].
Ten-eleven translocation family of dioxygenases (TETs) catalyse the successive oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) [101]. TET family contains three proteins TET1, TET2, and TET3 which can be detected in almost all tissues but are differentially expressed [102]. TET1 and TET2 are the main regulators of 5hmC levels in mouse embryonic stem cells (mESC) [103][104], while elsewhere TET2 and TET3 are expressed more ubiquitously [105]. Loss of TET activity and reduction of 5hmC is associated with a cancer phenotype [106]. Hydroxymethylation levels in mESC are high but decline after differentiation [107].
Histone modification is described by covalent post-translational modification of histones including methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation. They are also responsible for gene expression changes via modification of histone structure or disruption of transcription factor access to promoter sequences. Acetylation of histones is regulated by two main enzyme families; histone acetyltransferases (HATs) and histone deacetylases (HDAC) [108]. HDACs are essential for epigenetic regulation of gene expression, chromosome structure, and control of cellular stability. Their dysregulation is associated with loss of genomic integrity in cancer cells. HDACs remove the acetyl groups on histones which are themselves added by the histone acetyltransferases (HATs) [109]. Specific lysine residues on histones H2B, H3, and H4 are acetylated by HATs increasing DNA accessibility [110]. Histone methylation, a transfer of methyl groups onto constituent amino acids, occurs on the arginine, lysine and histidine residues on the histone proteins [111][112]. Three methylation forms are determined on histone lysine residues: mono-, di- and trimethylation and can be detected using selective antibodies that distinguish methylated histone residues. H3 Lys9 mono- and dimethylation are suggested to be relative to inactive genes in silent euchromatin domains with trimethylation at pericentric heterochromatin [113].
A further contributor to epigenetic modification is non-coding RNAs. These are a cluster of RNAs that regulate gene expression at the post-transcriptional level such as miRNAs (microRNAs), piRNAs (piwi-interacting RNAs), siRNAs (Small interfering RNAs), and lncRNAs (long non-coding RNAs). A number of studies have shown that non-coding RNAs also play an important role in the post-transcriptional regulation of epigenetic-modifying enzymes and genes involved in carcinogenesis [114]. lncRNAs play a role in transcriptional regulation via modification of chromatin structure and DNA methylation levels [88]. lncRNA localization to target loci and subsequent recruitment of chromatin-modifying protein factors can direct chromatin modification [115]. lncRNAs silence gene expression in processes such as X-inactivation or imprinting and also display an enhancer-like role in the activation of gene expression such as KLHL12 [116].
4. TERT Expression Regulation by Epigenetic Mechanisms
TERT is significant in carcinogenesis and, because of that, is a potential target for cancer treatment. However, the underlying epigenetic regulation of this gene remains ambiguous. DNA methylation, histone methylation–acetylation, and non-coding RNAs all play roles in the regulation of TERT expression in various biological processes including ageing and cancer [18]. The TERT promoter harbours a large CpG island (485 CpGs, −1800 to +2300 relative to ATG) within the promoter region, exons 1 and 2 (UCSC Genome Browser). Further, TERT promoter and exon 1 were found to be highly methylated in primary acute myeloid leukaemia cell lines [117][118]. DNA methylation is a genome-wide occurrence across CpG islands and non-coding regions, including promoters and enhancers. The promoter regions of genes considered to be highly expressed in cancer are predominantly hypomethylated [119]. The TERT promoter is an exception to this model due to its hypermethylated promoter pattern found in most tumour cells and accompanying robust expression [17].
Epigenetic and genetic factors both have roles in determining TERT expression levels in tumour cells. For instance, screening of 31 cancer types, including tumours and non-neoplastic tissue samples, revealed that 95% of them displayed telomerase activity and of that 53% displayed altered TERT promoter methylation, 31% TERT promoter mutations, 3% TERT amplification, 3% TERT structural variants, and 5% TERT promoter structural variants. In addition, TERT promoter methylation and mutations were associated with relative telomere shortening when compared to other alterations [15]. Structural 5p15.33 rearrangements, which occur in high-risk neuroblastomas, position the TERT coding sequence adjacent to strong enhancer elements. They are associated with histone modifications, methylation of the rearranged region (including the TERT locus) and high levels of TERT expression [120]. TERT promoter rearrangements could potentially change the position of enhancer elements which regulate activation of TERT expression [15].
Epigenetic mechanisms may be responsible for reversible silencing of the TERT during differentiation and stem cell research has the potential to contribute to our knowledge of down-regulation of TERT and other related gene regulations [69][121] Retinoic acid treatment of human embryonal carcinoma and human promyelocytic leukemia cell lines resulted in a decline in telomerase activity during differentiation [122]. hESC differentiation is accompanied by decreased expression of DNMT3A, DNMT3B, and TERT [123]. These indicate that epigenetic regulators, DNMTs and histone methyltransferases could play an important role in the regulation of TERT during differentiation. hESCs provide a vital model for the assessment of endogenous epigenetic gene regulation during differentiation in a non-transformed background. Embryonic differentiation contributes to telomeric attrition and the initiation of aging, however, mechanisms underlying telomerase down-regulation remain elusive. Epigenetic studies in ESC differentiation could indicate whether DNA methylation or histone modification contribute to this process and might also help our understanding of TERT regulation in telomerase-positive cancer cells.
References
- Muller, H.J. The Remaking of Chromosomes. Collect. Net. 1938, 13, 181–198.
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282.
- Blackburn, E.H.; Gall, J.G. A Tandemly Repeated Sequence at the Termini of the Extrachromosomal Ribosomal RNA Genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53.
- Greider, C.W.; Blackburn, E.H. Identification of a Specific Telomere Terminal Transferase Activity in Tetrahymena Extracts. Cell 1985, 43, 405–413.
- Morin, G.B. The Human Telomere Terminal Transferase Enzyme Is a Ribonucleoprotein That Synthesizes TTAGGG Repeats. Cell 1989, 59, 521–529.
- Harrington, L.; McPhail, T.; Mar, V.; Zhou, W.; Oulton, R.; Bass, M.B.; Arruda, I.; Robinson, M.O. A Mammalian Telomerase-Associated Protein. Science 1997, 275, 973–977.
- Holt, S.E.; Aisner, D.L.; Baur, J.; Tesmer, V.M.; Dy, M.; Ouellette, M.; Trager, J.B.; Morin, G.B.; Toft, D.O.; Shay, J.W.; et al. Functional Requirement of P23 and Hsp90 in Telomerase Complexes. Genes Dev. 1999, 13, 817–826.
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase Catalytic Subunit Homologs from Fission Yeast and Human. Science 1997, 277, 955–959.
- Autexier, C.; Lue, N.F. The Structure and Function of Telomerase Reverse Transcriptase. Annu. Rev. Biochem. 2006, 75, 493–517.
- Chai, W.; Ford, L.P.; Lenertz, L.; Wright, W.E.; Shay, J.W. Human Ku70/80 Associates Physically with Telomerase through Interaction with HTERT. J. Biol. Chem. 2002, 277, 47242–47247.
- Ting, N.S.Y.; Yu, Y.; Pohorelic, B.; Lees-Miller, S.P.; Beattie, T.L. Human Ku70/80 Interacts Directly with HTR, the RNA Component of Human Telomerase. Nucleic Acids Res. 2005, 33, 2090–2098.
- Avilion, A.A.; Piatyszek, M.A.; Gupta, J.; Shay, J.W.; Bacchetti, S.; Greider, C.W. Human Telomerase RNA and Telomerase Activity in Immortal Cell Lines and Tumor Tissues. Cancer Res. 1996, 56, 645–650.
- Ito, H.; Kyo, S.; Kanaya, T.; Takakura, M.; Koshida, K.; Namiki, M.; Inoue, M.; Gynecology, O. Detection of Human Telomerase Reverse Transcriptase Messenger RNA in Voided Urine Samples as a Useful Diagnostic Tool for Bladder Cancer. Clin. Cancer Res. 1998, 4, 2807–2810.
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013, 339, 957–959.
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic Analysis of Telomere Length and Somatic Alterations in 31 Cancer Types. Nat. Genet. 2017, 49, 349–357.
- Kawashima, M.; Kojima, M.; Ueda, Y.; Kurihara, S.; Hiyama, E. Telomere Biology Including TERT Rearrangements in Neuroblastoma: A Useful Indicator for Surgical Treatments. J. Pediatr. Surg. 2016, 51, 2080–2085.
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the Human Telomerase Catalytic Subunit (HTERT) Gene Correlates with Telomerase Activity. Int. J. Cancer 2002, 101, 335–341.
- Lewis, K.A.; Tollefsbol, T.O. Regulation of the Telomerase Reverse Transcriptase Subunit through Epigenetic Mechanisms. Front. Genet. 2016, 7, 1–12.
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425.
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.D.; Stadler, G.; Mender, I.; Lai, T.P.; Zhang, N.; Wright, W.E.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect—Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14.
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of Human Telomerase Reverse Transcriptase (HTERT) Regulation: Clinical Impacts in Cancer. J. Biomed. Sci. 2018, 25, 22.
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A Highly Conserved Repetitive DNA Sequence, (TTAGGG)n, Present at the Telomeres of Human Chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626.
- Greider, C.W. Telomeres Do D-Loop-T-Loop. Cell. 1999, 97, 419–422.
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; De Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514.
- De Lange, T. Shelterin: The Protein Complex That Shapes and Safeguards Human Telomeres. In Genes and Development; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2005; pp. 2100–2110.
- Rodriguez, R.; Müller, S.; Yeoman, J.A.; Trentesaux, C.; Riou, J.-F.; Balasubramanian, S. A Novel Small Molecule That Alters Shelterin Integrity and Triggers a DNA-Damage Response at Telomeres. J. Am. Chem. Soc. 2008, 130, 15758–15759.
- Jones, M.; Bisht, K.; Savage, S.A.; Nandakumar, J.; Keegan, C.E.; Maillard, I. The Shelterin Complex and Hematopoiesis. J. Clin. Investig. 2016, 1621–1629.
- Palm, W.; de Lange, T. How Shelterin Protects Mammalian Telomeres. Annu. Rev. Genet. 2008, 42, 301–334.
- Klapper, W.; Parwaresch, R.; Krupp, G. Telomere Biology in Human Aging and Aging Syndromes. Mech. Ageing Dev. 2001, 122, 695–712.
- Takubo, K.; Izumiyama-Shimomura, N.; Honma, N.; Sawabe, M.; Arai, T.; Kato, M.; Oshimura, M.; Nakamura, K.-I. Telomere Lengths Are Characteristic in Each Human Individual. Exp. Gerontol. 2002, 37, 523–531.
- Lingner, J.; Cooper, J.P.; Cech, T.R. Telomerase and DNA End Replication: No Longer a Lagging Strand Problem? Science 1995, 269, 1533–1534.
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636.
- Allday, M.J.; Inman, G.J.; Crawford, D.H.; Farrell, P.J. DNA Damage in Human B Cells Can Induce Apoptosis, Proceeding from G1/S When P53 Is Transactivation Competent and G2/M When It Is Transactivation Defective. EMBO J. 1995, 14, 4994–5005.
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM Kinase by Ionizing Radiation and Phosphorylation of P53. Science 1998, 281, 1677–1679.
- Shapiro, G.I.; Edwards, C.D.; Ewen, M.E.; Rollins, B.J. P16 INK4A Participates in a G 1 Arrest Checkpoint in Response to DNA Damage. Mol. Cell. Biol. 1998, 18, 378–387.
- Janknecht, R. On the Road to Immortality: HTERT Upregulation in Cancer Cells. FEBS Lett. 2004, 564, 9–13.
- Januszkiewicz, D.; Wysoki, J.; Lewandowski, K.; Pernak, M.; Nowicka, K.; Rembowska, J.; Nowak, J. Lack of Correlation between Telomere Length and Telomerase Activity and Expression in Leukemic Cells. Int. J. Mol. Med. 2003, 12, 935–938.
- Hornsby, P.J. Telomerase and the Aging Process. Exp. Gerontol. 2007, 42, 575–581.
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015.
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere Elongation in Immortal Human Cells without Detectable Telomerase Activity. EMBO J. 1995, 14, 4240–4248.
- Bailey, S.M.; Brenneman, M.A.; Goodwin, E.H. Frequent Recombination in Telomeric DNA May Extend the Proliferative Life of Telomerase-Negative Cells. Nucleic Acids Res. 2004, 32, 3743–3751.
- Grobelny, J.V.; Godwin, A.K.; Broccoli, D. ALT-Associated PML Bodies Are Present in Viable Cells and Are Enriched in Cells in the G2/M Phase of the Cell Cycle. J. Cell Sci. 2000, 113, 4577–4585.
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-Negative Immortalized Human Cells Contain a Novel Type of Promyelocytic Leukemia (PML) Body. Cancer Res. 1999, 59, 4175–4179.
- Liu, L.; Bailey, S.M.; Okuka, M.; Muñoz, P.; Li, C.; Zhou, L.; Wu, C.; Czerwiec, E.; Sandler, L.; Seyfang, A.; et al. Telomere Lengthening Early in Development. Nat. Cell Biol. 2007, 9, 1436–1441.
- Shay, J.W.; Wright, W.E. Role of Telomeres and Telomerase in Cancer. Semin. Cancer Biol. 2011, 21, 349–353.
- Leem, S.H.; Londoño-Vallejo, J.A.; Kim, J.H.; Bui, H.; Tubacher, E.; Solomon, G.; Park, J.E.; Horikawa, I.; Kouprina, N.; Barrett, J.C.; et al. The Human Telomerase Gene: Complete Genomic Sequence and Analysis of Tandem Repeat Polymorphisms in Intronic Regions. Oncogene 2002, 21, 769–777.
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere Position Effect: Regulation of Gene Expression with Progressive Telomere Shortening over Long Distances. Genes Dev. 2014, 28, 2464–2476.
- Kim, W.; Shay, J.W. Long-Range Telomere Regulation of Gene Expression: Telomere Looping and Telomere Position Effect over Long Distances (TPE-OLD). Differentiation 2018, 99, 1–9.
- Horikawa, I.; Cable, P.L.; Afshari, C.; Barrett, J.C. Cloning and Characterization of the Promoter Region of Human Telomerase Reverse Transcriptase Gene. Cancer Res. 1999, 59, 826–830.
- Cong, Y.S.; Wen, J.; Bacchetti, S. The Human Telomerase Catalytic Subunit HTERT: Organization of the Gene and Characterization of the Promoter. Hum. Mol. Genet. 1999, 8, 137–142.
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (HTERT) Gene. Genes 2016, 7, 50.
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad Network and the Transcriptional Control of Cell Behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699.
- Kyo, S.; Takakura, M.; Taira, T.; Kanaya, T.; Itoh, H.; Yutsudo, M.; Ariga, H.; Inoue, M. Sp1 Cooperates with C-Myc to Activate Transcription of the Human Telomerase Reverse Transcriptase Gene (HTERT). Nucleic Acids Res. 2000, 28, 669–677.
- Yu, J.; Yuan, X.; Sjöholm, L.; Liu, T.; Kong, F.; Ekström, T.J.; Björkholm, M.; Xu, D. Telomerase Reverse Transcriptase Regulates DNMT3B Expression/Aberrant DNA Methylation Phenotype and AKT Activation in Hepatocellular Carcinoma. Cancer Lett. 2018, 434, 33–41.
- Won, J.; Yim, J.; Kim, T.K. Sp1 and Sp3 Recruit Histone Deacetylase to Repress Transcription of Human Telomerase Reverse Transcriptase (HTERT) Promoter in Normal Human Somatic Cells. J. Biol. Chem. 2002, 277, 38230–38238.
- Oh, S.; Song, Y.H.; Yim, J.; Kim, T.K. Identification of Mad as a Repressor of the Human Telomerase (HTERT) Gene. Oncogene 2000, 19, 1485–1490.
- Wang, X.H.; Liu, B.R.; Qu, B.; Xing, H.; Gao, S.L.; Yin, J.M.; Wang, X.F.; Cheng, Y.Q. Silencing STAT3 May Inhibit Cell Growth through Regulating Signaling Pathway, Telomerase, Cell Cycle, Apoptosis and Angiogenesis in Hepatocellular Carcinoma: Potential Uses for Gene Therapy. Neoplasma 2011, 58, 158–164.
- Chung, S.S.; Aroh, C.; Vadgama, J.V. Constitutive Activation of STAT3 Signaling Regulates HTERT and Promotes Stem Cell-like Traits in Human Breast Cancer Cells. PLoS ONE 2013, 8, 83971.
- Wang, Y.Y.; Sun, G.; Luo, H.; Wang, X.F.; Lan, F.M.; Yue, X.; Fu, L.S.; Pu, P.Y.; Kang, C.S.; Liu, N.; et al. Mir-21 Modulates Htert through a Stat3-Dependent Manner on Glioblastoma Cell Growth. CNS Neurosci. Ther. 2012, 18, 722–728.
- Yamada, O.; Ozaki, K.; Akiyama, M.; Kawauchi, K. JAK-STAT and JAK-PI3K-MTORC1 Pathways Regulate Telomerase Transcriptionally and Posttranslationally in ATL Cells. Mol. Cancer Ther. 2012, 11, 1112–1121.
- Yamada, O.; Ozaki, K.; Furukawa, T.; Machida, M.; Wang, Y.H.; Motoji, T.; Mitsuishi, T.; Akiyama, M.; Yamada, H.; Kawauchi, K.; et al. Activation of STAT5 Confers Imatinib Resistance on Leukemic Cells through the Transcription of TERT and MDR1. Cell. Signal. 2011, 23, 1119–1127.
- Sinha-Datta, U.; Horikawa, I.; Michishita, E.; Datta, A.; Sigler-Nicot, J.C.; Brown, M.; Kazanji, M.; Barrett, J.C.; Nicot, C. Transcriptional Activation of HTERT through the NF-ΚB Pathway in HTLV-I-Transformed Cells. Blood 2004, 104, 2523–2531.
- Kyo, S.; Takakura, M.; Kanaya, T.; Zhuo, W.; Fujimoto, K.; Nishio, Y.; Orimo, A.; Inoue, M. Estrogen Activates Telomerase. Cancer Res. 1999, 59, 5917–5921.
- Misiti, S.; Nanni, S.; Fontemaggi, G.; Cong, Y.-S.; Wen, J.; Hirte, H.W.; Piaggio, G.; Sacchi, A.; Pontecorvi, A.; Bacchetti, S.; et al. Induction of HTERT Expression and Telomerase Activity by Estrogens in Human Ovary Epithelium Cells. Mol. Cell. Biol. 2000, 20, 3764–3771.
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. N. Engl. J. Med. 2011, 365, 537–547.
- Nishi, H.; Nakada, T.; Kyo, S.; Inoue, M.; Shay, J.W.; Isaka, K. Hypoxia-Inducible Factor 1 Mediates Upregulation of Telomerase (HTERT). Mol. Cell. Biol. 2004, 24, 6076–6083.
- Kyo, S.; Takakura, M.; Fujiwara, T.; Inoue, M. Understanding and Exploiting HTERT Promoter Regulation for Diagnosis and Treatment of Human Cancers. Cancer Sci. 2008, 99, 1528–1538.
- Lou, F.; Chen, X.; Jalink, M.; Zhu, Q.; Ge, N.; Zhao, S.; Fang, X.; Fan, Y.; Bjö, M.; Liu, Z.; et al. The Opposing Effect of Hypoxia-Inducible Factor-2A on Expression of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2007.
- Lai, S.R.; Phipps, S.; Liu, L.; Andrews, L.; Tollefsbol, T. Epigenetic Control of Telomerase and Modes of Telomere Maintenance in Aging and Abnormal Systems. Front. Biosci. 2005, 5, 1779–1796.
- Shats, I.; Milyavsky, M.; Tang, X.; Stambolsky, P.; Erez, N.; Brosh, R.; Kogan, I.; Braunstein, I.; Tzukerman, M.; Ginsberg, D.; et al. P53-Dependent down-Regulation of Telomerase Is Mediated by P21 Waf1. J. Biol. Chem. 2004, 279, 50976–50985.
- Kanaya, T.; Kyo, S.; Hamada, K.; Takakura, M.; Kitagawa, Y.; Harada, H.; Inoue, M. Adenoviral Expression of P53 Represses Telomerase Activity through Down-Regulation of Human Telomerase Reverse Transcriptase Transcription. Clin. Cancer Res. 2000, 6, 1239–1247.
- Takakura, M.; Kyo, S.; Kanaya, T.; Hirano, H.; Takeda, J.; Yutsudo, M.; Inoue, M. Cloning of Human Telomerase Catalytic Subunit (HTERT) Gene Promoter and Identification of Proximal Core Promoter Sequences Essential for Transcriptional Activation in Immortalized and Cancer Cells. Cancer Res. 1999, 59, 551–557.
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961.
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. Cancer. TERT Promoter Mutations and Telomerase Reactivation in Urothelial Cancer. Science 2015, 347, 1006–1010.
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 2017, 23, 703–713.
- Stern, J.L.; Theodorescu, D.; Vogelstein, B.; Papadopoulos, N.; Cech, T.R. Mutation of the TERT Promoter, Switch to Active Chromatin, and Monoallelic TERT Expression in Multiple Cancers. Genes Dev. 2015, 29, 2219–2224.
- Li, G.; Shen, J.; Cao, J.; Zhou, G.; Lei, T.; Sun, Y.; Gao, H.; Ding, Y.; Xu, W.; Zhan, Z.; et al. Alternative Splicing of Human Telomerase Reverse Transcriptase in Gliomas and Its Modulation Mediated by CX-5461. J. Exp. Clin. Cancer Res. 2018, 37.
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Tissue-Specific Alternate Splicing of Human Telomerase Reverse Transcriptase (HTERT) Influences Telomere Lengths during Human Development. Int. J. Cancer 2001, 91, 644–649.
- Hrdlickova, R.; Nehyba, J.; Bose, H.R. Alternatively Spliced Telomerase Reverse Transcriptase Variants Lacking Telomerase Activity Stimulate Cell Proliferation. Mol. Cell. Biol. 2012, 32, 4283–4296.
- Wong, M.S.; Wright, W.E.; Shay, J.W. Alternative Splicing Regulation of Telomerase: A New Paradigm? Trends Genet. 2014, 30, 430–438.
- Yi, X. Quantitation of Telomerase Components and HTERT MRNA Splicing Patterns in Immortal Human Cells. Nucleic Acids Res. 2001, 29, 4818–4825.
- Yi, X.; White, D.M.; Aisner, D.L.; Baur, J.A.; Wright, W.E.; Shay, J.W. An Alternate Splicing, Variant of the Human Telomerase Catalytic Subunit Inhibits Telomerase Activity. Neoplasia 2000, 2, 433–440.
- Colgin, L.M.; Wilkinson, C.; Englezou, A.; Kilian, A.; Robinson, M.O.; Reddel, R.R. The HTERTα Splice Variant Is a Dominant Negative Inhibitor of Telomerase Activity. Neoplasia 2000, 2, 426–432.
- Ulaner, G.A.; Hu, J.-F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Telomerase Activity in Human Development Is Regulated by Human Telomerase Reverse Transcriptase (HTERT) Transcription and by Alternate Splicing of HTERT Transcripts1. Cancer Res. 1998, 58, 4168–4172.
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative Splicing of HTERT Pre-MRNA: A Potential Strategy for the Regulation of Telomerase Activity. Int. J. Mol. Sci. 2017, 18, 567.
- Waddington, C.H. The Epigenotype. Int. J. Epidemiol. 2012, 41, 10–13.
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36.
- Mattick, J.S.; Makunin, I.V. Non-Coding RNA. Hum. Mol. Genet. 2006, 15 (Suppl. 1), R17–R29.
- Jones, P.A.; Laird, P.W. Cancer-Epigenetics Comes of Age. Nat. Genet. 1999, 21, 163–167.
- Feil, R.; Fraga, M.F. Epigenetics and the Environment: Emerging Patterns and Implications. Nat. Rev. Genet. 2012, 13, 97–109.
- Huang, K.; Shen, Y.; Xue, Z.; Bibikova, M.; April, C.; Liu, Z.; Cheng, L.; Nagy, A.; Pellegrini, M.; Fan, J.-B.; et al. A Panel of CpG Methylation Sites Distinguishes Human Embryonic Stem Cells and Induced Pluripotent Stem Cells. Stem Cell Rep. 2014, 2, 36–43.
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398.
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38.
- Ting, A.H.; Jair, K.; Schuebel, K.E.; Baylin, S.B. Differential Requirement for DNA Methyltransferase 1 in Maintaining Human Cancer Cell Gene Promoter Hypermethylation. Cancer Res. 2006, 66, 729–735.
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(Cytosine-C5)-Methyltransferase Methylates DNA Processively with High Preference for Hemimethylated Target Sites. J. Biol. Chem. 2004, 279, 48350–48359.
- Li, E. Chromatin Modification and Epigenetic Reprogramming in Mammalian Development. Nat. Rev. Genet. 2002, 3, 662–673.
- Beaulieu, N.; Morin, S.; Chute, I.C.; Robert, M.-F.; Nguyen, H.; MacLeod, A.R. An Essential Role for DNA Methyltransferase DNMT3B in Cancer Cell Survival. J. Biol. Chem. 2002, 277, 28176–28181.
- Ostler, K.R.; Davis, E.M.; Payne, S.L.; Gosalia, B.B.; Expósito-Céspedes, J.; Beau, M.M.L.; Godley, L.A. Cancer Cells Express Aberrant DNMT3B Transcripts Encoding Truncated Proteins. Oncogene 2007, 26, 5553–5563.
- Li, J.-Y.; Pu, M.-T.; Hirasawa, R.; Li, B.-Z.; Huang, Y.-N.; Zeng, R.; Jing, N.-H.; Chen, T.; Li, E.; Sasaki, H.; et al. Synergistic Function of DNA Methyltransferases Dnmt3a and Dnmt3b in the Methylation of Oct4 and Nanog. Mol. Cell. Biol. 2007, 27, 8748–8759.
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted Disruption of DNMT1, DNMT3A and DNMT3B in Human Embryonic Stem Cells. Nat. Genet. 2015, 47, 469–478.
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303.
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue Distribution of 5-Hydroxymethylcytosine and Search for Active Demethylation Intermediates. PLoS ONE 2010, 5, e15367.
- Huang, Y.; Chavez, L.; Chang, X.; Wang, X.; Pastor, W.A.; Kang, J.; Zepeda-Martínez, J.A.; Pape, U.J.; Jacobsen, S.E.; Peters, B.; et al. Distinct Roles of the Methylcytosine Oxidases Tet1 and Tet2 in Mouse Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1361–1366.
- Ito, S.; Dalessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet Proteins in 5mC to 5hmC Conversion, ES-Cell Self-Renewal and Inner Cell Mass Specification. Nature 2010, 466, 1129–1133.
- Tsagaratou, A.; González-Avalos, E.; Rautio, S.; Scott-Browne, J.P.; Togher, S.; Pastor, W.A.; Rothenberg, E.V.; Chavez, L.; Lähdesmäki, H.; Rao, A. TET Proteins Regulate the Lineage Specification and TCR-Mediated Expansion of INKT Cells. Nat. Immunol. 2017, 18, 45–53.
- Ficz, G.; Gribben, J.G. Loss of 5-Hydroxymethylcytosine in Cancer: Cause or Consequence? Genomics 2014, 104, 352–357.
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic Regulation of 5-Hydroxymethylcytosine in Mouse ES Cells and during Differentiation. Nature 2011, 473, 398–404.
- Xhemalce, B.; Dawson, M.A.; Bannister, A.J. Histone Modifications. In Encyclopedia of Molecular Cell Biology and Molecular Medicine; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011.
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395.
- Ng, S.S.; Yue, W.W.; Oppermann, U.; Klose, R.J. Dynamic Protein Methylation in Chromatin Biology. Cell. Mol. Life Sci. 2009, 66, 407–422.
- Bedford, M.T.; Clarke, S.G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13.
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol. Cell 2003, 12, 1591–1598.
- Guil, S.; Esteller, M. DNA Methylomes, Histone Codes and MiRNAs: Tying It All Together. Int. J. Biochem. Cell Biol. 2009, 87–95.
- Whitehead, J.; Pandey, G.K.; Kanduri, C. Regulation of the Mammalian Epigenome by Long Noncoding RNAs. Biochim. Biophys. Acta Gen. Subj. 2009, 936–947.
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long Noncoding RNAs with Enhancer-like Function in Human Cells. Cell 2010, 143, 46–58.
- Zhao, X.; Tian, X.; Kajigaya, S.; Cantilena, C.R.; Strickland, S.; Savani, B.N.; Mohan, S.; Feng, X.; Keyvanfar, K.; Dunavin, N.; et al. Epigenetic Landscape of the TERT Promoter: A Potential Biomarker for High Risk AML/MDS. Br. J. Haematol. 2016, 175, 427–439.
- Stern, J.L.; Paucek, R.D.; Huang, F.W.; Ghandi, M.; Nwumeh, R.; Costello, J.C.; Cech, T.R. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. 2017, 21, 3700–3707.
- Hanada, M.; Delia, D.; Aiello, A.; Stadtmauer, E.; Reed, J.C. Bcl-2 Gene Hypomethylation and High-Level Expression in B-Cell Chronic Lymphocytic Leukemia. Blood 1993, 82, 1820–1828.
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase Activation by Genomic Rearrangements in High-Risk Neuroblastoma. Nature 2015, 526, 700–704.
- Eitsuka, T.; Nakagawa, K.; Kato, S.; Ito, J.; Otoki, Y.; Takasu, S.; Shimizu, N.; Takahashi, T.; Miyazawa, T. Modulation of Telomerase Activity in Cancer Cells by Dietary Compounds: A Review. Int. J. Mol. Sci. 2018, 19, 478.
- Albanell, J.; Han, W.; Mellado, B.; Gunawardane, R.; Scher, H.I.; Dmitrovsky, E.; Moore, M.A. Telomerase Activity Is Repressed during Differentiation of Maturation-Sensitive but Not Resistant Human Tumor Cell Lines. Cancer Res. 1996, 56, 1503–1508.
- Phipps, S.M.O.; Love, W.K.; Mott, T.E.; Andrews, L.G.; Tollefsbol, T.O. Differential Expression of Epigenetic Modulators during Human Embryonic Stem Cell Differentiation. Mol. Biotechnol. 2009, 41, 201–207.

