 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Slawomir J. Terlikowski | + 1136 word(s) | 1136 | 2021-04-14 10:56:18 | | | |
| 2 | Vicky Zhou | Meta information modification | 1136 | 2021-04-28 11:48:48 | | |
Video Upload Options
Chimeric antigen receptors (CARs) are recombinant antigen receptors located on T lymphocytes or other immune cells that redirect their specificity and functions.
1. Introduction
The moieties used to bind to antigen fall in three general categories: (a) single-chain variable fragment (scFv) derived from antibodies; (b) antigen-binding fragment (Fab) selected from libraries or (c) nature ligands that engage their cognate receptor. The main rationale behind the use of CAR receptors in cancer immunotherapy is the rapid production of tumour-targeting T cells, bypassing the barriers and incremental kinetics of active immunisation [1]. The CAR-modified T cells acquire unique properties and act as ‘living drugs’ that may result in short-term, as well as long-term effects [2]. There are four generations of CARs used in clinical practice. The core structure of all four generations is an extracellular antigen recognition region with scFv, which is responsible for immunogenicity, affinity and specificity [3]. With scFvs, CARs can target specific cells and trigger downstream signals. Fragments of scFvs derive from an antigen-specific monoclonal antibody (mAb) [4]. The receptor’s extracellular domain originates from a cluster of differentiation CD4 and CD8. The transmembrane domain is usually derived from CD8, CD3-Ϛ (zeta), CD28 and intracellular tail including members of the tumour necrosis factor (TNF) receptor family, 4-1BB (CD137), OX-40 and CD27, has been incorporated to second and third generation [5]. The fourth generation of CARs is also called TRUCK T cells and was engineered to induce cytokines production, for example, IL-2, IL-12, IL-15 or granulocyte-macrophage colony-stimulating factor (GM-CSF) [6]. The green fluorescent protein (GFP) is a protein that exhibits bright green fluorescence when exposed to light in the blue to ultraviolet range. It can be added to every generation of CAR in term to estimate its specificity to bind target antigen via fluorescence microscope. Figure 1 represents the structure of CARs.
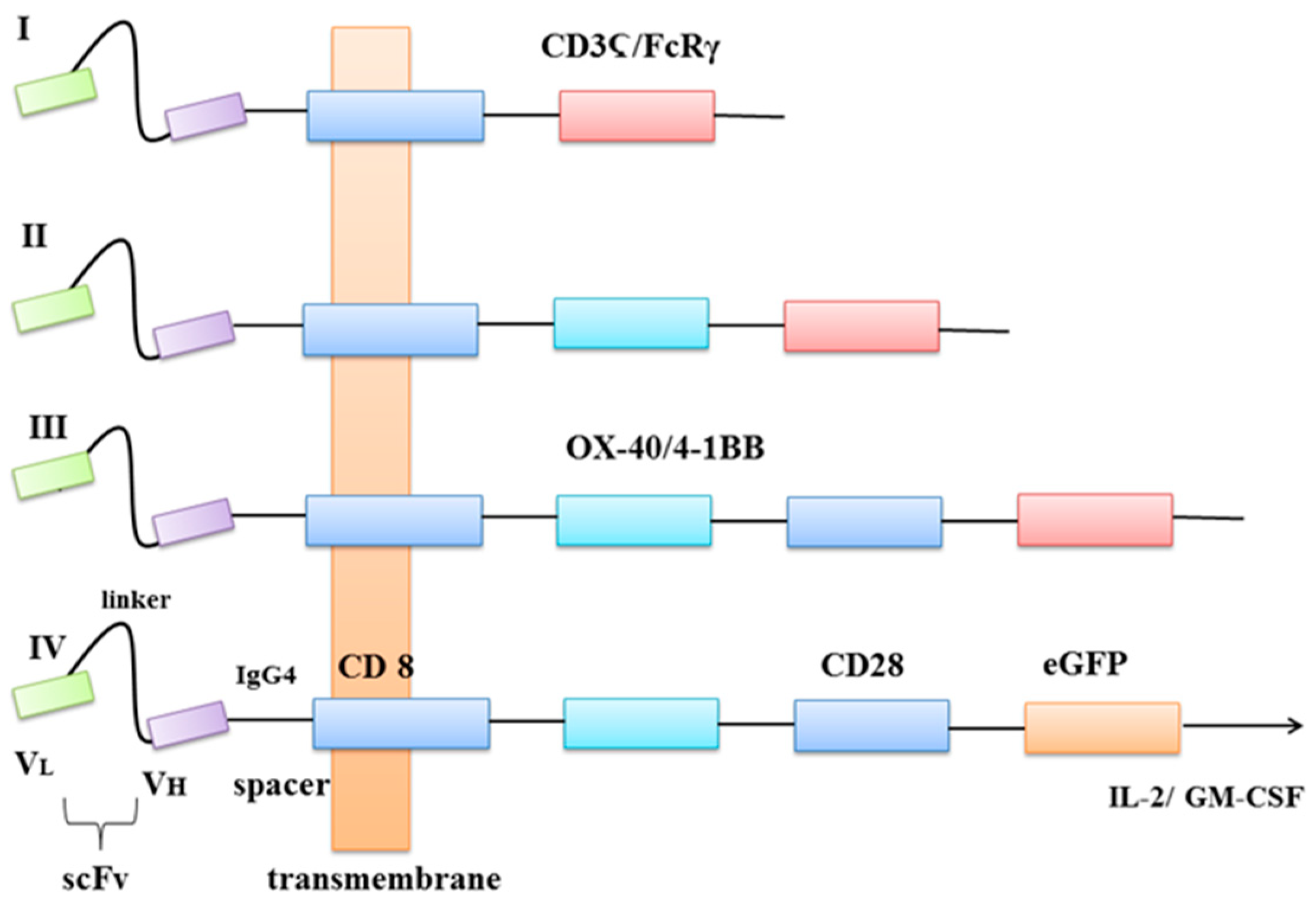
Figure 1. Four generations of CARs. VL—light chain variable domain, VH—heavy chain variable domain, scFv—a single-chain variable fragment, spacer—protein fragments fused together, CD8—transmembrane protein, OX-40—also known as CD134 glycoprotein receptor, tumour necrosis factor receptor superfamily, 4-1BB—glycoprotein receptor tumour necrosis factor receptor superfamily, CD3Ϛ—protein complex and T-cell co-receptor that is involved in activating both the cytotoxic T cell and T helper cells, FcRγ—receptor for inducing phagocytosis, CD28—a protein that provides costimulatory signals, eGFP—enhanced green fluorescent protein, Il-2—interleukin 2 (cytokine), GM-CSF—granulocyte-macrophage colony-stimulating factor.
Eshhar et al. designed structures that specifically recognise and respond to the antigen without signalisation of major histocompatibility complex (MHC) [7]. Unfortunately, first-generation CARs proved to be of limited clinical benefit because of failure in directing T-cell expansion upon repeated exposure to the antigen [8]. The 4-1BB ligand, CD137L is found on APCs (antigen-presenting cells) and binds to the 4-1BB superfamily, which is expressed on activated T Lymphocytes [9]. Savoldo et al. proposed incorporation of one stimulatory domain CD28 or 41BB to the second-generation CARs [10]. Third-generation CARs were formed by the incorporation of two or more costimulatory domains. On the other hand, their clinical effect in comparison to second-generation remains controversial [11][12]. The fourth-generation was developed to redirect T cells for universal cytokine killing, via the addition of an IL-12 expression cassette. IL-12 can accumulate in the target tissue and recruit a second wave of immune cells, e.g., NK cells, macrophages [13][14].
2. How Are CARs Engineered?
Scientists use several gene transfer methods to insert a specific gene into mice or human T lymphocytes. These methods differ in the expression levels and stability of mentioned CAR-T cells. In general, there are two main approaches in immune engineering: viral and non-viral [15]. Viral vectors have high infection rates; however, their production is costly and laborious. Moreover, there are also other challenges related to immunogenicity, carcinogenicity, low target cell specificity and inability to transfer large size genes. On the other hand, non-viral vectors can be relatively easy and cost-effectively produced. They are safe, can transfer large size genes and are less toxic. Their main disadvantages are low transfection efficiency and poor transgene expression [16]. Having considered the above, in this group, only the Sleeping Beauty (SB) transposon/transposase system with clustered regularly interspaced short palindromic repeats (CRISP/Cas9) has great potential [17]. Table 1 below lists the characteristics of different engineering methods of CARs.
Table 1. The characteristic of different engineering methods of CARs.
| Genetic Approach | Methods | Structure | Study | Target | Advantages | Disadvantages | Source |
|---|---|---|---|---|---|---|---|
| Viral | Retroviral vectors | ssRNA | in vivo | only mitosis | substitutability↑ | insertional mutagenesis↑ titre vector production ↑ |
[18] |
| Lentiviral vectors | ssRNA | in vivo | entire cycle | integration ↑ risk of insertional mutagenesis ↓ |
possible insertional mutagenesis↑ presence of regulatory proteins in the packaging construct transient expression of the transgene with integration-defective vector↑ |
[19] | |
| Adenoviruses | dsDNA | in vivo | entire cycle | toxicity↓ | integrity↓ | [20] | |
| Adeno-associated viral vectors | ssDNA | in vivo | entire cycle | infection efficiency ↑ gene expression ↑ |
internalisation ↓ endosomal trafficking↓ nuclear import↓ |
[21] | |
| Nonviral | Liposome-mediated gene transfer | lipid n-layer | in vitro | entire cycle | condensation of DNA ↑ infection efficiency ↑ |
transfection efficiency↓ | [22] |
| Messenger RNA-mediated gene transduction | ssRNA | in vitro | entire cycle | insertional mutations ↓ potential malignant transformation/ genotoxicity ↓ off-tumour, on-target side effects ↓ |
instable, non-biocompatible↓ low biodegradability, low efficacy↓ toxicity at high dose, difficult preparation, low transformation efficiency↓ |
[14][23] | |
| Sleeping Beauty transposon/transposase system | plasmid-plasmid | in vivo | entire cycle | integration ↑ | insertional mutagenesis ↓ | [18] |
A CAR intervention example of a mechanism in patients is shown in Figure 2.
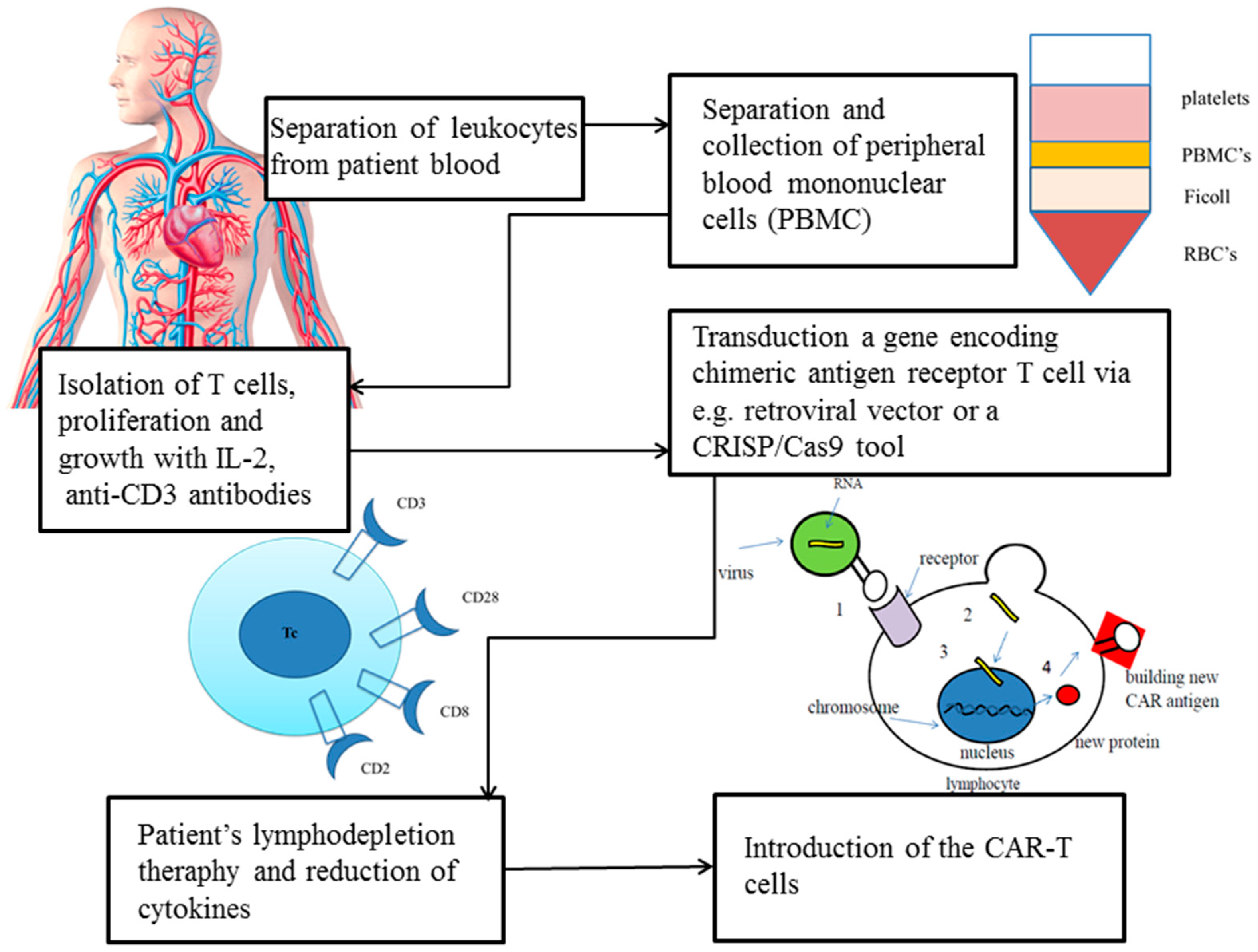
Figure 2. Transduction of a gene encoding a CAR in T cell via a retroviral vector or a CRISP/Cas9 tool [24].
The SB transposon system requires only two components: transposon DNA and transposase enzyme [25]. The most efficient way to deliver selected components into the target cell is the classical two-plasmid configuration: one for SB transposase and other for an artificial transposon flanked via terminal inverted repeats (TIR) on both sides of a vector [26]. This system also takes into account the origin of replication component and antibiotic resistance gene of choice. To setup transposon into the target cell, transfection or electroporation can be used [25]. To eliminate toxicity effect and decrease the immunogenic reaction of DNA transfection, it is best to use the current state-of-the-art delivery methods, messengers mRNA or minicircle DNA (MC) [27]. The SB’s production disadvantages are poor protein stability, low solubility and aggregation properties; however, incorporation of two mutations I212S and C176S into SB100X transposase improves these features [28]. A recent study indicates that using a catalytically inactive Cas9 (dCas9) with single-guide RNA approach may facilitate genetic material insertion into a genome [29]. Cas9 is a dual RNA-guided DNA endonuclease enzyme that uses base pairing to recognise and cleave target DNA with complementarity to the guide RNA, such as invading bacteriophage DNA or plasmid DNA [30]. Tethering the transposase toward a target that is overexpressed in the human genome dramatically increases the number of possible attach points and thus induces chances of targeted transposition with a flexible and easy-to-use RNA-guided system [31]. Pilot studies have indicated that SB is a safe and effective tool to manufacture therapeutic CAR-T cells in cancers [32][33][34][35].
References
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398.
- Wu, Y.; Jiang, S.; Ying, T. From Therapeutic Antibodies to Chimeric Antigen Receptors (CARs): Making Better CARs Based on Antigen-binding Domain. Expert Opin. Biol. Ther. 2016, 16, 1469–1478.
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors is Decisive for in vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135.
- Janda, A.; Eryilmaz, E.; Nakouzi, A.; Cowburn, D.; Casadevall, A. Variable Region Identical Immunoglobulins Differing in Isotype Express Different Paratopes. J. Biol. Chem. 2012, 287, 35409–35417.
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric Receptors with 4-1BB Signaling Capacity Provoke Potent Cytotoxicity Against Acute Lymphoblastic Leukemia. Leukemia 2004, 18, 676–684.
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T Cells Enhance Antitumor Efficacy and Overcome the Tumor Microenvironment. Sci. Rep. 2017, 7, 10541.
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724.
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115.
- O’Neill, R.E.; Du, W.; Mohammadpour, H.; Alqassim, E.; Qiu, J.; Chen, G.; McCarthy, P.L.; Lee, K.P.; Cao, X. T Cell-Derived CD70 Delivers an Immune Checkpoint Function in Inflammatory T Cell Responses. J. Immunol. 2017, 199, 3700–3710.
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 Costimulation Improves Expansion and Persistence of Chimeric Antigen Receptor-modified T Cells in Lymphoma Patients. J. Clin. Invest. 2011, 121, 1822–1826.
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific Adoptive Immunotherapy for Lymphoma Using a Chimeric Antigen Receptor with Both CD28 and 4-1BB Domains: Pilot Clinical Trial Results. Blood 2012, 119, 3940–3950.
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; FitzGerald, D.J.; Barrett, D.M.; et al. Anti-CD22-chimeric Antigen Receptors Targeting B-cell Precursor Acute Lymphoblastic Leukemia. Blood 2013, 121, 1165–1174.
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered with an Inducible Cytokine to Modulate the Tumor Stroma. Immunol. Rev. 2014, 257, 83–90.
- Rajan, T.S.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21, 6514.
- Pang, Y.; Hou, X.; Yang, C.; Liu, Y.; Jiang, G. Advances on Chimeric Antigen Receptor-modified T-cell Therapy for Oncotherapy. Mol. Cancer. 2018, 17, 91.
- Zhou, H.S.; Liu, D.P.; Liang, C.C. Challenges and Strategies: The Immune Responses in Gene Therapy. Med. Res. Rev. 2004, 24, 748–761.
- Aronovich, E.L.; McIvor, R.S.; Hackett, P.B. The Sleeping Beauty Transposon System: A Non-viral Vector for Gene Therapy. Hum. Mol. Genet. 2011, 20, R14–R20.
- Link, C.J., Jr.; Moorman, D.; Seregina, T.; Levy, J.P.; Schabold, K.J. A Phase I Trial of in vivo Gene Therapy with the Herpes Simplex Thymidine Kinase/Ganciclovir System for the Treatment of Refractory or Recurrent Ovarian Cancer. Hum. Gene Ther. 1996, 7, 1161–1179.
- Hu, W.S.; Pathak, V.K. Design of Retroviral Vectors and Helper Cells for Gene Therapy. Pharmacol. Rev. 2000, 52, 493–511.
- Li, Q.; Kay, M.A.; Finegold, M.; Stratford-Perricaudet, L.D.; Woo, S.L. Assessment of Recombinant Adenoviral Vectors for Hepatic Gene Therapy. Hum. Gene Ther. 1993, 4, 403–409.
- Isayeva, T.; Ren, C.; Ponnazhagan, S. Recombinant Adeno-associated Virus 2-mediated Antiangiogenic Prevention in a Mouse Model of Intraperitoneal Ovarian Cancer. Clin. Cancer Res. 2005, 11, 1342–1347.
- Fraley, R.; Subramani, S.; Berg, P.; Papahadjopoulos, D. Introduction of Liposome-encapsulated SV40 DNA into Cells. J. Biol. Chem. 1980, 255, 10431–10435.
- Zhao, Y.; Zheng, Z.; Cohen, C.J.; Gattinoni, L.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. High-efficiency Transfection of Primary Human and Mouse T Lymphocytes Using RNA Electroporation. Mol. Ther. 2006, 151–159.
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical Development of CAR T Cells-challenges and Opportunities in Translating Innovative Treatment Concepts. EMBO Mol. Med. 2017, 9, 1183–1197.
- Hamm, A.; Krott, N.; Breibach, I.; Blindt, R.; Bosserhoff, A.K. Efficient Transfection Method for Primary Cells. Tissue Eng. 2002, 8, 235–245.
- Stevens, J.M.; Galyov, E.E.; Stevens, M.P. Actin-dependent Movement of Bacterial Pathogens. Nat. Rev. Microbiol. 2006, 4, 91–101.
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell Engineering Using Non-viral Sleeping Beauty Transposition from Minicircle Vectors. Leukemia 2017, 31, 186–194.
- Amberger, M.; Ivics, Z. Latest Advances for the Sleeping Beauty Transposon System: 23 Years of Insomnia but Prettier than Ever: Refinement and Recent Innovations of the Sleeping Beauty Transposon System Enabling Novel, Nonviral Genetic Engineering Applications. Bioessays 2020, 42, e2000136.
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821.
- Kovač, A.; Miskey, C.; Menzel, M.; Grueso, E.; Gogol-Döring, A.; Ivics, Z. RNA-guided Retargeting of Sleeping Beauty Transposition in Human Cells. Elife 2020, 9, e53868.
- Belur, L.R.; Podetz-Pedersen, K.M.; Sorenson, B.S.; Hsu, A.H.; Parker, J.B.; Carlson, C.S.; Saltzman, D.A.; Ramakrishnan, S.; McIvor, R.S. Inhibition of Angiogenesis and Suppression of Colorectal Cancer Metastatic to the Liver Using the Sleeping Beauty Transposon System. Mol. Cancer 2011, 10, 14.
- Magnani, C.F.; Turazzi, N.; Benedicenti, F.; Calabria, A.; Tenderini, E.; Tettamanti, S.; Giordano Attianese, G.M.; Cooper, L.J.; Aiuti, A.; Montini, E.; et al. Immunotherapy of Acute Leukemia by Chimeric Antigen Receptor-modified Lymphocytes Using an Improved Sleeping Beauty Transposon Platform. Oncotarget 2016, 7, 51581–51597.
- Ohlfest, J.R.; Demorest, Z.L.; Motooka, Y.; Vengco, I.; Oh, S.; Chen, E.; Scappaticci, F.A.; Saplis, R.J.; Ekker, S.C.; Low, W.C.; et al. Combinatorial Antiangiogenic Gene Therapy by Nonviral Gene Transfer Using the Sleeping Beauty Transposon Causes Tumor Regression and Improves Survival in Mice Bearing Intracranial Human Glioblastoma. Mol. Ther. 2005, 12, 778–788.
- Xu, X.; Qiu, J.; Sun, Y. The Basics of CAR T Design and Challenges in Immunotherapy of Solid Tumors—Ovarian Cancer as a Model. Hum. Vaccin. Immunother. 2017, 13, 1548–1555.

