 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Agnes BOULLIER | + 3091 word(s) | 3091 | 2021-01-06 06:51:56 | | | |
| 2 | Lily Guo | -5 word(s) | 3086 | 2021-04-29 04:40:52 | | |
Video Upload Options
Over the last few years, preclinical and clinical studies have emphasized the role of extracellular vesicles (EVs) in human diseases. These particles are delimited by a lipid bilayer and are released by almost all cell types and in all organisms. EVs appear to have biological effects in various pathophysiological situations and especially in renal disease. In human organs, EVs can interact with cells and prompt the release of many different molecules, such as proteins, lipids and nucleic acids, that, in turn, regulate various cell signaling pathways. Moreover, EVs are present in the urine and the blood and therefore can be used as potential diagnostic biomarkers in human diseases, such as chronic kidney disease (CKD, also known as chronic renal failure).
1. Introduction
1.1. Classification of EVs
In recent years, studies of the role of EVs in human disease have shown that these vesicles are involved in several physiological pathways. Firstly, EVs are involved in cell–cell communication because they deliver various bioactive molecules and components to recipient cells [1]. In fact, the vesicles carry proteins, lipids, nucleic acids and other metabolites that can have many different cellular and supracellular effects [1]. EVs are highly heterogeneous but can be divided into three main categories: exosomes (30–150 nm in diameter), microvesicles (also referred to as microparticles or ectosomes: 100–1000 nm) and apoptotic vesicles (50–5000 nm) [2]. The three subgroups differ with regard to their mode of biogenesis (Figure 1)
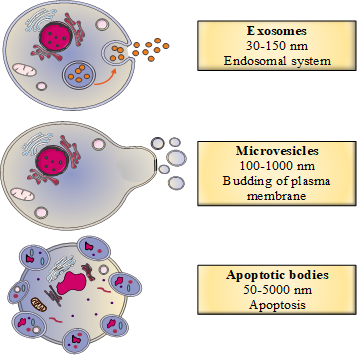
Figure 1. The classification of extracellular vesicles (EVs) as a function of their biogenesis.
1.2. Biogenesis of EVs
1.2.1. The Biogenesis of Exosomes
Exosomes are produced from a complex endosomal system involving two different pathways, one of which depends on a protein complex called the endosomal sorting complex, which is required for transport (ESCRT) [3]. Typically, the budding of late endosomes enables the production of intraluminal vesicles within multivesicular endosomes. These vesicles then fuse with the plasma membrane to give secreted exosomes [3].In the ESCRT-dependent pathway, four protein complexes (ESCRT-0, I, II, III) bind to the hexameric AAA ATPase Vps4 complex, which provides the energy for exosome biogenesis [3]. The intraluminal vesicles are formed because of the negative curvature of the endosomal membrane, which depends on recognition of ubiquitinated endosomal proteins by ESCRT-0 [4]. Next, ESCRT-I, II and III interact with ESCRT-0 to enable intraluminal vesicle formation in multivesicular endosomes [4]. Lastly, the AAA ATPase Vsp4 complex causes ESCRT-III to separate from the multivesicular endosomal membrane, and the ESCRT complex is recycled [4].Several studies have highlighted the existence of an ESCRT-independent pathway for exosome biogenesis, based on lipid raft microdomains located in the late endosome membrane [2]. These microdomains contain cone-shaped molecules (e.g., lysobisphosphatidic acid and ceramides) that can induce negative curvature of the endosomal membrane [4][5].
1.2.2. The Biogenesis of Microvesicles
In contrast to exosomes, microvesicles are generated by direct budding from the plasma membrane. During microvesicle biogenesis, the interaction between the cytoskeleton and the plasma membrane weakens and several proteins (such as calpains or lipid translocases) are activated [2]. This cytoskeletal remodeling has been linked to an increase in the intracytosolic calcium concentration, which, in turn, induces phosphatidylserine externalization, bud formation and microvesicle secretion [6].
1.2.3. The Biogenesis of Apoptotic Bodies
Unlike exosomes and microvesicles (which are secreted during normal cellular processes), apoptotic bodies are only released during apoptosis [2]. First, the cell membrane buds. Next, externalization of phosphatidylserine (as observed during microvesicle biogenesis) leads to cytoskeletal remodeling and then the formation of apoptotic bodies [2].
1.3. Secretion of EVs
Several different molecules are involved in the secretion of EVs in general and exosomes in particular, since microvesicles and apoptotic bodies are generated directly by the budding of the plasma membrane.The Ras-related proteins in brain (Rab) family is composed of more than 60 GTPases [7]. Screening studies have identified a number of small GTPases proteins involved in exosome secretion in various cell-based models [7]. Rab GTPases are involved in (i) the movement of multivesicular endosomes from the cytoplasm to the plasma membrane and (ii) the fusion of the cell’s plasma membrane with that of exosome-containing multivesicular endosomes [7].Other molecules are involved in exosome secretion [7]. Thus, soluble n-ethylmaleimide-sensitive-factor attachment protein receptor complexes are thought to be involved in exosome secretion in various cell types [7]. The interaction between soluble n-ethylmaleimide-sensitive factor attachment protein and its receptor leads to the fusion of the plasma membrane with the multivesicular endosome membrane [7]. Some studies have identified other molecules with a potential role in exosome secretion, including diacyl glycerol kinase α, which appears to downregulate multivesicular endosome formation [8]. Citron kinase (a RhoA effector) and the V0 subunit of V-ATPase appear to be also involved in exosome secretion [9][10].It is noteworthy that the exosome secretion pathway depends on the EVs’ cellular origin.
1.4. Fate of EVs
The release of EVs from parental cells may interact with target cells and influence target cell behavior and phenotype behavior [11]. Indeed, EVs carry bioactive molecules such as proteins, lipids and nucleic acids, which have been shown to impact target cells via several mechanisms: (1) direct stimulation of the target cells upon binding to cell surface; (2) transfer of activated receptors to recipient cells; and (3) epigenetic reprogramming of recipient cells via the delivery of functional protein, lipids and RNA.After EVs are released, they can interact directly with recipient cells by binding to cell surface integrins, proteoglycans or extracellular matrix components, thus inducing different biological processes [1]. For instance, intracellular adhesion molecule-1 on exosomes can interact with lymphocyte function-associated antigen-1 on dendritic cells [12]. Similarly, milk fat globule-EGF factor 8 protein (a lactadherin precursor present on immune cells) can interact with the phosphatidylserine on exosomes [12]. These interactions allow exosome internalization, the presentation of exosome-derived peptides to T cells and T cell activation [12]. Several studies have identified other molecular interactions between EVs and recipient cells in various cellular models [7].The EV membrane can also merge with the plasma membrane of recipient cells, thus releasing the EVs’ contents (miRNAs, proteins, peptides, nucleic acids, etc.) into the recipient’s cytoplasm [1]. EVs are particularly enriched with miRNAs loaded into EVs via RNA binding protein recruitment [13]. For instance, synaptotagmin-binding cytoplasmic RNA-interaction protein (SYNCRIP) can be associated with miR-3470a and miR-194-2-3p [13]. Other RNA binding proteins are implicated in the sorting process for miRNAs into EVs, such as argonaute2 protein (Ago2) or Y-box binding protein 1 (YBX-1) [13]. The uptake of EVs by the recipient cell occurs through a variety of processes, such as micropinocytosis, endocytosis and phagocytosis (for a review, see References [1][14]). After release, the EVs’ contents can induce various biological processes [7]. For instance, some EVs are enriched in enzymes such as cyclooxygenase and thromboxane synthase, which can regulate platelet activation and aggregation by metabolizing arachidonic acid into thromboxane [15].After their release, EVs can also be found in various biological fluids, such as blood or urine. Although the half-life of EVs has not been determined, EVs appear to be stable in biological fluids for at least several hours [7]. Moreover, intravenously injected EVs can be found in many organs (such as the spleen, lung and liver) [7].
2. Extracellular Vesicles in a Pathological Setting: A Focus on CKD
2.1. EVs and CKD
Although EVs appear to be associated with many diseases, their involvement in renal diseases (e.g., CKD) is especially strong. Furthermore, depending on their cell origin, EVs are linked to various pathophysiological processes in CKD.
Firstly, many clinical studies have shown that CKD patients have abnormally high levels of EVs in general and endothelial microvesicles in particular. Indeed, endothelial microvesicles are found to be higher in many cohorts of patients with end-stage renal disease (ESRD) [16][17][18][19][20][21]. Furthermore, it is known that plasma endothelial microvesicles are strong predictors of cardiovascular diseases. As such, they can be used as markers of endothelial dysfunction, atherosclerosis and arterial stiffness in CKD patients [16]. Moreover, the level of endothelial microvesicles in haemodialyzed patients is inversely correlated with laminar shear stress, which is a major determinant of plasma endothelial microvesicle levels in ESRD [22].
Other studies found that levels of platelet-derived microvesicles were also elevated in CKD patients [23] and that these EVs appear to have procoagulant and prothrombotic activity [24][25]. Circulating levels of microvesicles were also significantly elevated in CKD patients with coronary artery disease [26]. Furthermore, Benito-Martin et al. reported on elevated urine levels of exosomal proteins in CKD patients [27].
2.2. Effect of Haemodialysis on EVs
CKD progression leads to ESRD, which requires renal replacement therapy. Several dialysis techniques are currently used, including haemodialysis (HD), haemofiltration (HF), haemodiafiltration, and peritoneal dialysis (PD). HD is the most common renal replacement therapy for CKD patients. It effectively removes free small molecular weight compounds but not protein-bound molecules or large molecules. The results of the few studies of the effects of HD on EVs are subject to debate. Indeed, some studies found that plasma EV levels are higher in haemodialyzed patients than in healthy subjects [18][20][28][29]. It is noteworthy that the types of altered microvesicle vary from one study to another. In contrast, some studies observed a fall in the level of EVs during HD [30][31]. By analyzing protein markers, Ruzicka et al. showed that the observed reduction in circulating EVs was due to adsorption of the EVs to the dialysis membrane rather than ultrafiltration [31]. Moreover, Cavallari et al. showed that online haemodiafiltration is better than HD because it decreased cardiomortality [32], inflammation [33] and miR223 expression in EVs, thus reducing VSMCs calcification and improving angiogenesis by human umbilical vein endothelial cells (HUVECs) [34]. In conclusion, the choice of the method for renal replacement therapy is critical for EVs.
2.3. Impact of Uremic Toxins on EVs
In addition to traditional cardiovascular risk factors, CKD patients also have non-traditional CKD-specific cardiovascular risk factors, such as the accumulation of uremic toxins. Indeed, as kidney function decreases, uremic toxins accumulate in the body fluids of CKD patients. Many studies have reported abnormally high concentrations of uremic toxins in the serum of CKD patients [35]. EVs appear to be linked to the physiopathology of CKD. Indeed, several studies have highlighted a link between the accumulation of uremic toxins in CKD patients and EV release [36]. Usually, EV secretion is elevated under uremic conditions. Thus, the level of platelet-derived microvesicles was higher in a cohort of 18 uremic patients than in a group of healthy subjects [37]. Gao et al. also reported that uremic patients had an elevated number of circulating microvesicles derived from peripheral blood cells and endothelial cells [38].
3. Extracellular Vesicles as Biomarkers of CKD
Conventional biomarkers for CKD diagnosis include a low GFR and albuminuria [39]. Recent studies have focused on urinary EVs since they may be non-invasive diagnostic or prognostic biomarkers [40].EVs in urine are of great interest as renal disease markers because they reflect the nephron’s physiopathological status [41][42][43][44][45][46][47][48]. Indeed, Pisitkun et al.’s proteomic analysis showed that most EVs in urine come from glomerular and tubular cells because circulating EVs cannot cross the filtration barrier under physiological conditions [41]. Lv et al. suggested that urinary exosomes containing the inflammatory chemokine CCL2 mRNA are biomarkers of CKD [49]. Indeed, elevated levels of these exosomes were observed in the kidney and urine of CKD rats and patients with IgA nephropathy [49]. Exosomes can transfer CCL2 mRNA from tubular epithelial cells to macrophages, promoting inflammatory kidney injury [49]. Furthermore, Lv et al. showed that mRNA encoding CD2AP in urinary exosomes was correlated with kidney function and renal fibrosis [50].Clinical and preclinical studies have highlighted the value of miRs (transported by EVs) as CKD biomarkers [51][52][53][54][55][56][57][58][59][60][61][62]. Indeed, exosomes of patients with kidney diseases contain high levels of miR [55]. Liu et al. have shown that urinary miR-126 (probably secreted by exosomes) is more strongly expressed in patients with diabetic nephropathy, whereas treated patients have lower expression levels of this miR [57]. A specific urinary exosomal miR profile was found in patients with focal segmental glomerulosclerosis, a disease that can lead to CKD [59]. Furthermore, miR29 and miR200 are downregulated in urinary exosomes from CKD patients [62]. The miR-451-5p and miR-16 found in urinary exosomes from diabetic rats also appear to protect the kidney tissue [61].Some researchers provide technical information about urinary EVs and miR transported by these EVs as non-invasive biomarkers for kidney diseases such as CKD [55][56]. However, it is important to note that EVs cannot always be handled easily and that the creation of standardized protocols is required if EVs are to be used as biomarkers in the clinic.
4. The Therapeutic Potential of Extracellular Vesicles in CKD
As described above, EVs are involved in the physiopathology of CKD, VC and uraemic conditions. Therefore, EVs are potential therapeutic targets that could be inhibited by various pharmacological agents [63][64]. EVs could also be used as drug delivery systems because of their involvement in cell–cell communication [65].
4.1. Inhibition of EVs
Several in vitro studies have demonstrated that various molecules can be used to block EV release or the uptake of EVs by recipient cells, as reviewed elsewhere [63][64].The complexity and heterogeneity of EV biogenesis complicates the development of drugs that inhibit EVs. In principle, inhibitors could act at several different stages of EV biogenesis. The Rab family is involved in exosome secretion [7]. It has been shown that the inhibition of Rab proteins (Rab27A and Rab27B) leads to a decrease in exosome secretion [66]. Several other inhibitors (including calpeptin, Y27632 and manumycin A) affect EV trafficking [64]. The best studied compound is calpeptin, a reversible, semi-synthetic peptidomimetic aldehyde inhibitor of calpains [64]. Calpains is a family of calcium-dependent cytosolic proteases the action of which on cytoskeletal remodeling promotes microvesicle shedding [2]. Therefore, the inhibition of calpains leads to a decrease in microvesicle release by cells [67].EV release can also be inhibited by blocking lipid metabolism [64]. As described above, lipid metabolism in general and ceramide metabolism in particular are involved in EV biogenesis [7]. One such inhibitor is panthetine. It inhibits cholesterol synthesis by changing the membrane fluidity and thus blocking the translocation of phosphatidylserine to the outer leaflet, an essential process in microvesicle production [64]. Sphingomyelinases are important enzymes for exosome and microvesicle formation and therefore constitute potential therapeutic targets [64]. Thus, treatment with imipramine or GW4869 (inhibitors of acid sphingomyelinase and membrane neutral sphingomyelinase, respectively) leads to a reduction in EV secretion [64]. Other inhibitors (such as antiplatelet molecules, antioxidants, statins, calcium-channel blockers and proton pump inhibitors) also reduce EV biogenesis (for detailed reviews, see References [63][64]).Another way of counteracting the EVs’ effects is to reduce their uptake by recipient cells [63]. Even though the inhibition of EV uptake increases the levels of circulating EVs, it will still reduce the harmful effects in recipient cells. This inhibition can be either achieved with antibodies targeting various molecules (e.g., Annexin V and integrin αvβ3) or with drugs that modify, for example, microfilament formation [63].These inhibitors must be used with caution, however. Special attention should be paid to their mechanism of action and side effects to ensure that these drugs do not reduce EV biogenesis in or uptake by healthy cells.
4.2. The Therapeutic Potential of MSC- and EPC-Derived EVs
Given that MSCs have beneficial effects in human diseases like CKD [68], MSC-derived EVs have attracted great interest for their therapeutic potential in CKD. Thus, a protective role for MSC EVs was found in patients with CKD stage 3–4, as evidenced by a higher estimated GFR and lower values for blood urea, serum creatinine and the urine albumin to creatinine ratio [69]. These effects were accompanied by an increase in the plasma levels of anti-inflammatory cytokines (such as TGF-β1 and interleukin-10) and a decrease in the plasma level of TNF-α [69]. Furthermore, MSC-derived microvesicles have been shown to protect rats against CKD and inhibit renal fibrosis [70]. Another in vivo study in mice with CKD induced by subtotal (5/6) nephrectomy showed that MSC-derived EVs were able to decrease not only proteinuria, serum creatinine and uric acid but also fibrosis, interstitial lymphocyte infiltration and tubular atrophy [71]. Kidney MSC microvesicles exert antifibrotic effects by decreasing the endothelial-to-mesenchymal transition and increasing TGF-β-induced HUVEC proliferation [72]. In vivo, the researchers obtained the same results in unilateral ureteral obstruction (UUO) mice and evidenced the inhibition of inflammatory cell infiltration and tubulointerstitial fibrosis [72]. The MSC microvesicles’ protective role was also observed in vitro for proximal tubular epithelial cells, with enhanced E-cadherin expression and lower α-smooth muscle actin (α-SMA) secretion; these results were confirmed in vivo in UUO mice [73]. Furthermore, MSC-derived exosomes containing miR-let7c induce antifibrotic effects, especially with a decrease in expression levels of collagen IVα1, α-SMA and TGF-β1 receptor in vitro [74]. MSC-derived EVs can also reduce renal inflammation and fibrosis and increase medullary oxygenation in pigs with metabolic syndrome and renal artery stenosis [75]. Interestingly, van Koppen et al. have shown that MSC-derived exosomes had no effect on CKD progression, systolic blood pressure and renal damage in 5/6-nephrectomized rats but did stimulate angiogenesis [76]. However, the conditioned, exosome-containing medium in the latter study had protective effects on induced CKD, hypertension and glomerular injury, suggesting that, along with exosomes, other molecules in the conditioned medium were needed to induce protective effects [76].Circulating endothelial progenitor cells (EPC) play an important role in the maintenance of vascular integrity and the regeneration of the vascular system. The number of circulating EPC in CKD patients is 30% lower than in healthy controls [77][78][79]. Surdacki et al. showed that EPC number is in fact inversely correlated to the degree of kidney dysfunction [80]. The low EPC count [81] and dysfunction [82] may contribute to an increased risk of CVD in CKD patients [83][84]. EPC therapy has been shown to decrease inflammation and proteinuria and to preserve kidney function [85]. This beneficial effect is thought to be mediated in part in a paracrine manner, and recent studies have shown that EPC-EVs may be responsible for this effect by transferring miRNA. Thus, EPC-EVs activate angiogenesis by the transfer of mRNA, especially miR126 and miR296, which have been shown to modulate proliferation, angiogenesis, apoptosis and inflammation [86][87][88]. EPC-EVs also protect against CKD progression after ischemia-reperfusion injury I by inhibiting, in particular, tubule interstitial fibrosis [88].
4.3. EVs as Drug Carriers
Thanks to their small size and low immunogenicity, EVs are able to transfer endogenous and exogenous drug compounds (such as siRNA [89], protein drugs or pharmaceutical drugs) into recipient cells. Moreover, the EVs’ lipid bilayer of membrane protects their contents from degradation. The advantages of EVs include their non-toxicity, their long half-life and, importantly, their ability to cross barriers such as the blood-brain barrier [90]. Specific binding of EVs to the target cells is important for effectiveness. To this end, donor cells can be genetically engineered so that a specific ligand is expressed on the EVs they release [91] and can be specifically recognized by the target recipient cells. Most studies have been performed in cell-based or animal models. Only a few drug-carrying EVs have been tested in the clinic [65].Despite the EVs’ potential as delivery systems, some important issues need to be addressed for clinical use; these include the culture conditions, EV purification/quantification and the choice of the donor cells.
References
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228.
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170.
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208.
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9.
- Matsuo, H.; Chevallier, J.; Mayran, N.; Blanc, I.L.; Ferguson, C.; Fauré, J.; Blanc, N.S.; Matile, S.; Dubochet, J.; Sadoul, R.; et al. Role of LBPA and Alix in Multivesicular Liposome Formation and Endosome Organization. Science 2004, 303, 531–534.
- Hugel, B.; Martínez, M.C.; Kunzelmann, C.; Freyssinet, J.-M. Membrane Microparticles: Two Sides of the Coin. Physiology 2005, 20, 22–27.
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289.
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase α regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ. 2011, 18, 1161–1173.
- Loomis, R.J.; Holmes, D.A.; Elms, A.; Solski, P.A.; Der, C.J.; Su, L. Citron Kinase, a RhoA Effector, Enhances HIV-1 Virion Production by Modulating Exocytosis. Traffic 2006, 7, 1643–1653.
- Liégeois, S.; Benedetto, A.; Garnier, J.-M.; Schwab, Y.; Labouesse, M. The V0-ATPase mediates apical secretion of exosomes containing Hedgehog-related proteins in Caenorhabditis elegans. J. Cell Biol. 2006, 173, 949–961.
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581.
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.G.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266.
- Groot, M.; Lee, H. Sorting Mechanisms for MicroRNAs into Extracellular Vesicles and Their Associated Diseases. Cells 2020, 9, 1044.
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3.
- Boilard, E. Extracellular vesicles and their content in bioactive lipid mediators: More than a sack of microRNA. J. Lipid Res. 2018, 59, 2037–2046.
- Dursun, I.; Poyrazoglu, H.M.; Gunduz, Z.; Ulger, H.; Yykylmaz, A.; Dusunsel, R.; Patyroglu, T.; Gurgoze, M. The relationship between circulating endothelial microparticles and arterial stiffness and atherosclerosis in children with chronic kidney disease. Nephrol. Dial. Transplant. 2009, 24, 2511–2518.
- Amabile, N.; Guérin, A.P.; Tedgui, A.; Boulanger, C.M.; London, G.M. Predictive value of circulating endothelial microparticles for cardiovascular mortality in end-stage renal failure: A pilot study. Nephrol. Dial. Transplant. 2012, 27, 1873–1880.
- Merino, A.; Portolés, J.; Selgas, R.; Ojeda, R.; Buendia, P.; Ocaña, J.; Bajo, M.A.; del Peso, G.; Carracedo, J.; Ramírez, R.; et al. Effect of different dialysis modalities on microinflammatory status and endothelial damage. Clin. J. Am. Soc. Nephrol. 2010, 5, 227–234.
- Amabile, N.; Guérin, A.P.; Leroyer, A.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating Endothelial Microparticles Are Associated with Vascular Dysfunction in Patients with End-Stage Renal Failure. JASN 2005, 16, 3381–3388.
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573.
- Trappenburg, M.C.; van Schilfgaarde, M.; Frerichs, F.C.P.; Spronk, H.M.H.; ten Cate, H.; de Fijter, C.W.H.; Terpstra, W.E.; Leyte, A. Chronic renal failure is accompanied by endothelial activation and a large increase in microparticle numbers with reduced procoagulant capacity. Nephrol. Dial. Transplant. 2012, 27, 1446–1453.
- Boulanger, C.M.; Amabile, N.; Guérin, A.P.; Pannier, B.; Leroyer, A.S.; Mallat, C.N.Z.; Tedgui, A.; London, G.M. In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 2007, 49, 902–908.
- Almquist, T.; Mobarrez, F.; Jacobson, S.H.; Wallén, H.; Hjemdahl, P. Effects of lipid-lowering treatment on circulating microparticles in patients with diabetes mellitus and chronic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 944–952.
- Ando, M.; Iwata, A.; Ozeki, Y.; Tsuchiya, K.; Akiba, T.; Nihei, H. Circulating platelet-derived microparticles with procoagulant activity may be a potential cause of thrombosis in uremic patients. Kidney Int. 2002, 62, 1757–1763.
- Burton, J.O.; Hamali, H.A.; Singh, R.; Abbasian, N.; Parsons, R.; Patel, A.K.; Goodall, A.H.; Brunskill, N.J. Elevated levels of procoagulant plasma microvesicles in dialysis patients. PLoS ONE 2013, 8, e72663.
- Chen, Y.-L.; Chen, C.-H.; Wallace, C.G.; Wang, H.-T.; Yang, C.-C.; Yip, H.-K. Levels of circulating microparticles in patients with chronic cardiorenal disease. J. Atheroscler. Thromb. 2015, 22, 247–256.
- Benito-Martin, A.; Ucero, A.C.; Zubiri, I.; Posada-Ayala, M.; Fernandez-Fernandez, B.; Cannata-Ortiz, P.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Egido, J.; Alvarez-Llamas, G.; et al. Osteoprotegerin in exosome-like vesicles from human cultured tubular cells and urine. PLoS ONE 2013, 8, e72387.
- Daniel, L.; Fakhouri, F.; Joly, D.; Mouthon, L.; Nusbaum, P.; Grunfeld, J.-P.; Schifferli, J.; Guillevin, L.; Lesavre, P.; Halbwachs-Mecarelli, L. Increase of circulating neutrophil and platelet microparticles during acute vasculitis and hemodialysis. Kidney Int. 2006, 69, 1416–1423.
- de Laval, P.; Mobarrez, F.; Almquist, T.; Vassil, L.; Fellström, B.; Soveri, I. Acute effects of haemodialysis on circulating microparticles. Clin. Kidney J. 2018, 12, 456–462.
- Georgatzakou, H.T.; Tzounakas, V.L.; Kriebardis, A.G.; Velentzas, A.D.; Kokkalis, A.C.; Antonelou, M.H.; Papassideri, I.S. Short-term effects of hemodiafiltration versus conventional hemodialysis on erythrocyte performance. Can. J. Physiol. Pharmacol. 2018, 96, 249–257.
- Ruzicka, M.; Xiao, F.; Abujrad, H.; Al-Rewashdy, Y.; Tang, V.A.; Langlois, M.-A.; Sorisky, A.; Ooi, T.C.; Burger, D. Effect of hemodialysis on extracellular vesicles and circulating submicron particles. BMC Nephrol. 2019, 20, 294.
- Grooteman, M.P.C.; van den Dorpel, M.A.; Bots, M.L.; Penne, E.L.; van der Weerd, N.C.; Mazairac, A.H.A.; den Hoedt, C.H.; van der Tweel, I.; Lévesque, R.; Nubé, M.J.; et al. Effect of online hemodiafiltration on all-cause mortality and cardiovascular outcomes. J. Am. Soc. Nephrol. 2012, 23, 1087–1096.
- den Hoedt, C.H.; Bots, M.L.; Grooteman, M.P.C.; van der Weerd, N.C.; Mazairac, A.H.A.; Penne, E.L.; Levesque, R.; ter Wee, P.M.; Nubé, M.J.; Blankestijn, P.J.; et al. Online hemodiafiltration reduces systemic inflammation compared to low-flux hemodialysis. Kidney Int. 2014, 86, 423–432.
- Cavallari, C.; Dellepiane, S.; Fonsato, V.; Medica, D.; Marengo, M.; Migliori, M.; Quercia, A.D.; Pitino, A.; Formica, M.; Panichi, V.; et al. Online Hemodiafiltration Inhibits Inflammation-Related Endothelial Dysfunction and Vascular Calcification of Uremic Patients Modulating miR-223 Expression in Plasma Extracellular Vesicles. J. Immunol. 2019, 202, 2372–2383.
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270.
- Favretto, G.; da Cunha, R.S.; Dalboni, M.A.; de Oliveira, R.B.; de Carvalho Barreto, F.; Massy, Z.A.; Stinghen, A.E.M. Endothelial Microparticles in Uremia: Biomarkers and Potential Therapeutic Targets. Toxins 2019, 11, 267.
- Nomura, S.; Shouzu, A.; Nishikawa, M.; Kokawa, T.; Yasunaga, K. Significance of Platelet-Derived Microparticles in Uremia. NEF 1993, 63, 485.
- Gao, C.; Xie, R.; Yu, C.; Ma, R.; Dong, W.; Meng, H.; Zhang, Y.; Si, Y.; Zhang, Z.; Novakovic, V.; et al. Thrombotic Role of Blood and Endothelial Cells in Uremia through Phosphatidylserine Exposure and Microparticle Release. PLoS ONE 2015, 10, e0142835.
- Zhong, J.; Yang, H.-C.; Fogo, A.B. A perspective on chronic kidney disease progression. Am. J. Physiol. Renal Physiol. 2017, 312, F375–F384.
- Erdbrügger, U.; Le, T.H. Extracellular Vesicles in Renal Diseases: More than Novel Biomarkers? J. Am. Soc. Nephrol. 2016, 27, 12–26.
- Pisitkun, T.; Shen, R.-F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373.
- Fang, D.Y.; King, H.W.; Li, J.Y.; Gleadle, J.M. Exosomes and the kidney: Blaming the messenger. Nephrology 2013, 18, 1–10.
- Dursun, I.; Yel, S.; Unsur, E. Dynamics of circulating microparticles in chronic kidney disease and transplantation: Is it really reliable marker? World J. Transplant. 2015, 5, 267–275.
- Gámez-Valero, A.; Lozano-Ramos, S.I.; Bancu, I.; Lauzurica-Valdemoros, R.; Borràs, F.E. Urinary extracellular vesicles as source of biomarkers in kidney diseases. Front. Immunol. 2015, 6, 6.
- Lin, J.; Li, J.; Huang, B.; Liu, J.; Chen, X.; Chen, X.-M.; Xu, Y.-M.; Huang, L.-F.; Wang, X.-Z. Exosomes: Novel biomarkers for clinical diagnosis. Sci. World J. 2015, 2015, 657086.
- Zhang, W.; Zhou, X.; Zhang, H.; Yao, Q.; Liu, Y.; Dong, Z. Extracellular vesicles in diagnosis and therapy of kidney diseases. Am. J. Physiol. Renal Physiol. 2016, 311, F844–F851.
- Lyu, L.-L.; Feng, Y.; Liu, B.-C. Urinary Biomarkers for Chronic Kidney Disease with a Focus on Gene Transcript. Chin. Med. J. 2017, 130, 2251–2256.
- Barreiro, K.; Holthofer, H. Urinary extracellular vesicles. A promising shortcut to novel biomarker discoveries. Cell Tissue Res. 2017, 369, 217–227.
- Lv, L.-L.; Feng, Y.; Wen, Y.; Wu, W.-J.; Ni, H.-F.; Li, Z.-L.; Zhou, L.-T.; Wang, B.; Zhang, J.-D.; Crowley, S.D.; et al. Exosomal CCL2 from Tubular Epithelial Cells Is Critical for Albumin-Induced Tubulointerstitial Inflammation. J. Am. Soc. Nephrol. 2018, 29, 919–935.
- Lv, L.-L.; Cao, Y.-H.; Pan, M.-M.; Liu, H.; Tang, R.-N.; Ma, K.-L.; Chen, P.-S.; Liu, B.-C. CD2AP mRNA in urinary exosome as biomarker of kidney disease. Clin. Chim. Acta 2014, 428, 26–31.
- Abbasian, N.; Herbert, K.E.; Pawluczyk, I.; Burton, J.O.; Bevington, A. Vesicles bearing gifts: The functional importance of micro-RNA transfer in extracellular vesicles in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2018, 315, F1430–F1443.
- Shang, F.; Wang, S.-C.; Hsu, C.-Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.-C.; Wang, Y.-T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. JASN 2017, 28, 3251–3261.
- Beltrami, C.; Clayton, A.; Phillips, A.O.; Fraser, D.J.; Bowen, T. Analysis of urinary microRNAs in chronic kidney disease. Biochem. Soc. Trans. 2012, 40, 875–879.
- Goettsch, C.; Hutcheson, J.D.; Aikawa, E. MicroRNA in cardiovascular calcification: Focus on targets and extracellular vesicle delivery mechanisms. Circ. Res. 2013, 112, 1073–1084.
- Lv, L.-L.; Cao, Y.; Liu, D.; Xu, M.; Liu, H.; Tang, R.-N.; Ma, K.-L.; Liu, B.-C. Isolation and quantification of microRNAs from urinary exosomes/microvesicles for biomarker discovery. Int. J. Biol. Sci. 2013, 9, 1021–1031.
- Cheng, L.; Sharples, R.A.; Scicluna, B.J.; Hill, A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles 2014, 3.
- Liu, Y.; Gao, G.; Yang, C.; Zhou, K.; Shen, B.; Liang, H.; Jiang, X. Stability of miR-126 in Urine and Its Potential as a Biomarker for Renal Endothelial Injury with Diabetic Nephropathy. Int. J. Endocrinol. 2014, 2014, 393109.
- Papadopoulos, T.; Belliere, J.; Bascands, J.-L.; Neau, E.; Klein, J.; Schanstra, J.P. miRNAs in urine: A mirror image of kidney disease? Expert Rev. Mol. Diagn. 2015, 15, 361–374.
- Ramezani, A.; Devaney, J.M.; Cohen, S.; Wing, M.R.; Scott, R.; Knoblach, S.; Singhal, R.; Howard, L.; Kopp, J.B.; Raj, D.S. Circulating and urinary microRNA profile in focal segmental glomerulosclerosis: A pilot study. Eur. J. Clin. Investig. 2015, 45, 394–404.
- Wang, B.; Zhang, C.; Zhang, A.; Cai, H.; Price, S.R.; Wang, X.H. MicroRNA-23a and MicroRNA-27a Mimic Exercise by Ameliorating CKD-Induced Muscle Atrophy. J. Am. Soc. Nephrol. 2017, 28, 2631–2640.
- Mohan, A.; Singh, R.S.; Kumari, M.; Garg, D.; Upadhyay, A.; Ecelbarger, C.M.; Tripathy, S.; Tiwari, S. Urinary Exosomal microRNA-451-5p Is a Potential Early Biomarker of Diabetic Nephropathy in Rats. PLoS ONE 2016, 11, e0154055.
- Lv, L.-L.; Cao, Y.-H.; Ni, H.-F.; Xu, M.; Liu, D.; Liu, H.; Chen, P.-S.; Liu, B.-C. MicroRNA-29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am. J. Physiol. Renal Physiol. 2013, 305, F1220–F1227.
- Karpman, D.; Ståhl, A.-L.; Arvidsson, I. Extracellular vesicles in renal disease. Nat. Rev. Nephrol. 2017, 13, 545–562.
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2019, 9.
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087.
- Datta, A.; Kim, H.; McGee, L.; Johnson, A.E.; Talwar, S.; Marugan, J.; Southall, N.; Hu, X.; Lal, M.; Mondal, D.; et al. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci. Rep. 2018, 8, 8161.
- Yano, Y.; Shiba, E.; Kambayashi, J.; Sakon, M.; Kawasaki, T.; Fujitani, K.; Kang, J.; Mori, T. The effects of calpeptin (a calpain specific inhibitor) on agonist induced microparticle formation from the platelet plasma membrane. Thromb. Res. 1993, 71, 385–396.
- Yun, C.W.; Lee, S.H. Potential and Therapeutic Efficacy of Cell-based Therapy Using Mesenchymal Stem Cells for Acute/chronic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1619.
- Nassar, W.; El-Ansary, M.; Sabry, D.; Mostafa, M.A.; Fayad, T.; Kotb, E.; Temraz, M.; Saad, A.-N.; Essa, W.; Adel, H. Umbilical cord mesenchymal stem cells derived extracellular vesicles can safely ameliorate the progression of chronic kidney diseases. Biomater. Res. 2016, 20, 21.
- Gatti, S.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Cantaluppi, V.; Tetta, C.; Camussi, G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol. Dial. Transplant. 2011, 26, 1474–1483.
- He, J.; Wang, Y.; Sun, S.; Yu, M.; Wang, C.; Pei, X.; Zhu, B.; Wu, J.; Zhao, W. Bone marrow stem cells-derived microvesicles protect against renal injury in the mouse remnant kidney model. Nephrology 2012, 17, 493–500.
- Choi, H.Y.; Lee, H.G.; Kim, B.S.; Ahn, S.H.; Jung, A.; Lee, M.; Lee, J.E.; Kim, H.J.; Ha, S.K.; Park, H.C. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem. Cell Res. Ther. 2015, 6, 18.
- He, J.; Wang, Y.; Lu, X.; Zhu, B.; Pei, X.; Wu, J.; Zhao, W. Micro-vesicles derived from bone marrow stem cells protect the kidney both in vivo and in vitro by microRNA-dependent repairing. Nephrology 2015, 20, 591–600.
- Wang, B.; Yao, K.; Huuskes, B.M.; Shen, H.-H.; Zhuang, J.; Godson, C.; Brennan, E.P.; Wilkinson-Berka, J.L.; Wise, A.F.; Ricardo, S.D. Mesenchymal Stem Cells Deliver Exogenous MicroRNA-let7c via Exosomes to Attenuate Renal Fibrosis. Mol. Ther. 2016, 24, 1290–1301.
- Eirin, A.; Zhu, X.-Y.; Puranik, A.S.; Tang, H.; McGurren, K.A.; van Wijnen, A.J.; Lerman, A.; Lerman, L.O. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. 2017, 92, 114–124.
- van Koppen, A.; Joles, J.A.; van Balkom, B.W.M.; Lim, S.K.; de Kleijn, D.; Giles, R.H.; Verhaar, M.C. Human embryonic mesenchymal stem cell-derived conditioned medium rescues kidney function in rats with established chronic kidney disease. PLoS ONE 2012, 7, e38746.
- de Groot, K.; Bahlmann, F.H.; Sowa, J.; Koenig, J.; Menne, J.; Haller, H.; Fliser, D. Uremia causes endothelial progenitor cell deficiency. Kidney Int. 2004, 66, 641–646.
- Choi, J.-H.; Kim, K.L.; Huh, W.; Kim, B.; Byun, J.; Suh, W.; Sung, J.; Jeon, E.-S.; Oh, H.-Y.; Kim, D.-K. Decreased number and impaired angiogenic function of endothelial progenitor cells in patients with chronic renal failure. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1246–1252.
- Ozkok, A.; Aktas, E.; Yilmaz, A.; Telci, A.; Oflaz, H.; Deniz, G.; Yildiz, A. Decrease in endothelial progenitor cells associated with inflammation, but not with endothelial dysfunction in chronic hemodialysis patients. Clin Nephrol 2013, 79, 21–30.
- Surdacki, A.; Marewicz, E.; Wieteska, E.; Szastak, G.; Rakowski, T.; Wieczorek-Surdacka, E.; Dudek, D.; Pryjma, J.; Dubiel, J.S. Association between endothelial progenitor cell depletion in blood and mild-to-moderate renal insufficiency in stable angina. Nephrol. Dial. Transplant. 2008, 23, 2265–2273.
- Maruyama, S.; Taguchi, A.; Iwashima, S.; Ozaki, T.; Yasuda, K.; Kikuchi-Taura, A.; Soma, T.; Ishii, H.; Murohara, T.; Takahashi, H.; et al. Low circulating CD34+ cell count is associated with poor prognosis in chronic hemodialysis patients. Kidney Int. 2008, 74, 1603–1609.
- Goligorsky, M.S.; Yasuda, K.; Ratliff, B. Dysfunctional endothelial progenitor cells in chronic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 911–919.
- Herbrig, K.; Pistrosch, F.; Oelschlaegel, U.; Wichmann, G.; Wagner, A.; Foerster, S.; Richter, S.; Gross, P.; Passauer, J. Increased total number but impaired migratory activity and adhesion of endothelial progenitor cells in patients on long-term hemodialysis. Am. J. Kidney Dis. 2004, 44, 840–849.
- Lin, C.-J.; Wu, C.-J.; Wu, P.-C.; Pan, C.-F.; Wang, T.-J.; Sun, F.-J.; Liu, H.-L.; Chen, H.-H.; Yeh, H.-I. Indoxyl Sulfate Impairs Endothelial Progenitor Cells and Might Contribute to Vascular Dysfunction in Patients with Chronic Kidney Disease. Kidney Blood Press Res. 2016, 41, 1025–1036.
- Sangidorj, O.; Yang, S.H.; Jang, H.R.; Lee, J.P.; Cha, R.; Kim, S.M.; Lim, C.S.; Kim, Y.S. Bone marrow-derived endothelial progenitor cells confer renal protection in a murine chronic renal failure model. Am. J. Physiol. Renal Physiol. 2010, 299, F325–F335.
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell–derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448.
- Cantaluppi, V.; Medica, D.; Mannari, C.; Stiaccini, G.; Figliolini, F.; Dellepiane, S.; Quercia, A.D.; Migliori, M.; Panichi, V.; Giovannini, L.; et al. Endothelial progenitor cell-derived extracellular vesicles protect from complement-mediated mesangial injury in experimental anti-Thy1.1 glomerulonephritis. Nephrol. Dial. Transplant. 2015, 30, 410–422.
- Cantaluppi, V.; Gatti, S.; Medica, D.; Figliolini, F.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia–reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012, 82, 412–427.
- Wahlgren, J.; Karlson, T.D.L.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130.
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345.
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191.

