 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Md. Imtaiyaz Hassan | + 2930 word(s) | 2930 | 2021-04-20 07:53:24 | | | |
| 2 | Karina Chen | Meta information modification | 2930 | 2021-04-28 05:03:01 | | |
Video Upload Options
Sphingolipid metabolites have emerged as critical players in the regulation of various physiological processes. Ceramide and sphingosine induce cell growth arrest and apoptosis, whereas sphingosine-1-phosphate (S1P) promotes cell proliferation and survival.
1. Sphingolipid Metabolism
Sphingolipid metabolism is a highly coordinated broad-spectrum cellular pathway, wherein several pathways are linked together. The synthesis and degradation of bioactive sphingolipids are regulated by several enzymes having fluxes of diverse metabolites [1]. Almost all the key enzymes of sphingolipid metabolic pathways have been identified which has unveiled the complexity of these pathways and their distinct subcellular compartmentalization [2][3]. An outline of the highly coordinated sphingolipid metabolism connecting several pathways is represented in Figure 1. The bioactive lipid ceramide occupies the central position in the sphingolipid metabolism hub and can be produced through diverse pathways via three primary mechanisms [4][5]. The first one is the de novo generation of ceramide that begins in the endoplasmic reticulum (ER) with 3-ketosphinganine from palmitoyl-CoA and serine units the catalytic action of serine palmitoyltransferase (SPT).
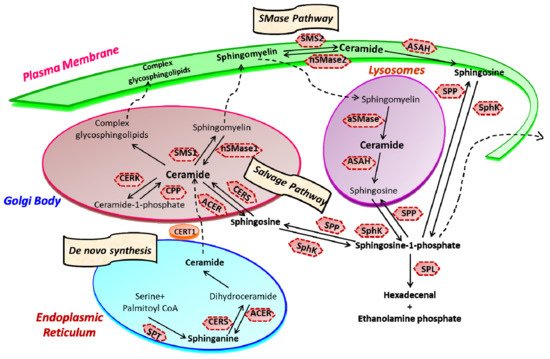
Figure 1. Sphingolipid metabolism and compartmentalization. Three significant mechanisms for ceramide generation are illustrated. Ceramide is synthesized via s “de novo pathway” in the ER from where it is transported to the Golgi bodies through CERT and serves as a substrate for the synthesis of complex glycosphingolipids (GLSL) and sphingomyelin (SM). GLSL and SM are transported to the plasma membrane through vesicular transport. In another mechanism; ceramide can be generated by the action of neutral or acidic SMases (“SMase pathway”) in the plasma membrane. Finally, in the “salvage pathway”, ceramide is synthesized from sphingosine released from the lysosome by the catalytic action of CERS. (SPT, Serine palmitoyltransferase; CERS, Ceramide synthase; CERK, Ceramide kinase; CPP, Ceramide-1-phosphate phosphatase; SMS, Sphingomyelin synthase; SMase, Sphingomyelinase; ACER, ceramidase in Golgi bodies; ASAH, ceramidase in lysosomes; SK, sphingosine kinase; SPP, Sphingosine-1-phosphate phosphatase; SPL, S1P lyase) (Figure is adapted from reference [6]).
Ceramide synthase (CERS) catalyzes the acylation of dihydrosphingosine to form dihydroceramide which is converted to ceramide by desaturase [7][8]. Thereafter, ceramide can follow multiple intracellular routes. Ceramide is transported to the Golgi bodies by ceramide transport protein (CERT) where it is metabolically converted to sphingomyelin and various complex sphingolipids [5][9]. The transport of ceramide from ER to the site of sphingomyelin synthesis occurs via both vesicular and non-vesicular pathways [10][11].
In Golgi bodies, ceramide is converted to sphingomyelin, a vital component of the plasma membrane, by incorporating phosphocholine head group by sphingomyelin synthases (SMS). Glycosylation of ceramide by glycosyl or galactosyl CERS results in the formation of complex glycosphingolipids, an integral part of cell membranes and known to confer drug resistance to cancerous cells [12][13]. Additionally, ceramide can be directly phosphorylated to form ceramide-1-phosphate (C1P) by the catalytic activity of a specific ceramide kinase (CERK), which plays a crucial role in regulating cell homeostasis as well as in mediating inflammatory responses. C1P is further transported to the plasma membrane or other organelles for various biological signaling cascades by the C1P transfer protein. Conversely, ceramide can be generated through the catabolic degradation of sphingomyelin via sphingomyelinase (SMase), another mechanism of ceramide generation [14]. Three distinct forms of mammalian SMase having different pH optima i.e., acidic, neutral, and alkaline, has been identified [15]. While neutral SMase is responsible for generating ceramide in Golgi bodies (nSMase1) and plasma membrane (nSmase2). The acid SMase leads to ceramide synthesis from sphingomyelin in lysosomes (aSMase) [16][17]. In addition to that, the alkaline SMase is involved in the breakdown of dietary sphingomyelin in the intestine.
Finally, the third mechanism is the salvage pathway through which ceramide is generated from sphingosine by the action of CERS. Ceramide can be metabolized back to bioactive lipid sphingosine by specific ceramidase (CDase or ACER in Golgi bodies) and ASAH in lysosomes). Subsequently, sphingosine kinase (SphK or SK) catalyzes the phosphorylation of sphingosine to form another potent signaling molecule, sphingosine-1-phosphate (S1P). The S1P can either be degraded to ethanolamine phosphate and fatty aldehyde by lyase (SPL) or dephosphorylated to sphingosine by S1P phosphatase and re-acylated back to ceramide [4][5][18][19][14].
Ceramide and S1P are among the key sphingolipids having special biological functions. They function oppositely, with ceramide acting as a pro-apoptotic molecule controlling growth arrest and autophagy, whereas S1P functions as a pro-survival molecule and regulates cell proliferation, survival, angiogenesis, and cell trafficking. The dynamic balance of these two opposite-acting sphingolipids is referred to as the “sphingolipid-rheostat” regulating the cell’s fate [20][21][22]. Under normal physiological conditions, the balance of the sphingolipid rheostat is well maintained by the functional activity of a key enzyme, SphK. The bending of this intricate balance towards sphingosine induces cell cycle arrest and apoptosis. In contrast, the accumulation of S1P inside activates cell growth and proliferation pathways, often leading to pathological conditions such as cancer and other inflammatory diseases. The notion that the “sphingolipid-rheostat” defines the cell fate by controlling the levels of ceramide and S1P is further strengthened by the fact that inhibition of SphK leads to decreased S1P, and enhanced ceramide, ensuring cell death.
One of the essential aspects of ceramide metabolism is the segregation of metabolic pools and bioactive metabolites within the cells, which seems crucial for ceramide to function as a regulatory signaling molecule. Such compartmentalization is facilitated by the distinct and well-defined subcellular localization of enzymes regulating ceramide metabolism [3][23][24]. Another key feature of ceramide metabolism is the translocation of ceramide from its synthesis i.e., the endoplasmic reticulum (ER) to the Golgi bodies, where it is transformed into sphingomyelin sphingosine and complex glycosphingolipids. Ceramide is converted to sphingosine by the catalytic activity of ceramidase (CDase) in the nucleus, plasma membrane, lysosomes, and mitochondria. Sphingosine is further converted to S1P by SphK1 in the cytoplasm and plasma membrane for intracellular and extracellular signaling, respectively [19][14][25]. The production of S1P in ER and mitochondria is facilitated by the catalytic action of SphK2, where it is localized [26][27].
2. Sphingosine Kinase
SphK is a lipid kinase that catalyzes the ATP-dependent phosphorylation of sphingosine to S1P [28][29]. Two isoforms of SphK—SphK1 and SphK2—have been characterized in humans that regulate various cellular processes [30][31]. They catalyze the same biochemical reaction but differ in their substrate affinities, tissue distribution, and subcellular localization [32]. SphK1 resides in the cytoplasm under normal physiological conditions, but it is translocated to the plasma membrane when activated. In contrast, SphK2 is present in the nucleus. Although the two isoenzymes are highly homologous, they have been observed to perform distinct functions. SphK1 shows pro-survival effects, whereas studies point towards a pro-apoptotic role of SphK2 [33][34][35].
2.1. SphK1 Activation and Functions
The function of SphK1 and subsequent intracellular levels of S1P is regulated by various mechanisms, including gene transcription, translational regulation and posttranslational modifications [36][37][38]. In particular, the activity of SphK1 is regulated by various agonists and stimuli that facilitate its phosphorylation and translocation to the plasma membrane, where it synthesizes S1P from sphingosine (Figure 2). A range of external stimuli that induce phosphorylation of SphK1 includes transforming growth factor β (TGF-β), tumor necrosis factor α (TNF-α), pro-inflammatory cytokine, and various growth factors via the activation of receptor tyrosine kinases, G-protein coupled receptors and toll-like receptors [39][40][41]. TGF-β has been observed to induce SphK1 activity by upregulating its gene expression levels [42][43][44]. Moreover, TGF-β exposure reduces the activity of S1P phosphatase, ultimately leading to a transient and rapid increase in S1P intracellular levels [45][46]. Another activator of SphK1 is TNFα that enhances SphK1 activity in the human umbilical vein endothelial cell (HUVEC) [47][48]. Other endogenous agonists include prolactin and 17β-estradiol, both upregulate the expression and activity of SphK1 [49][50][51].
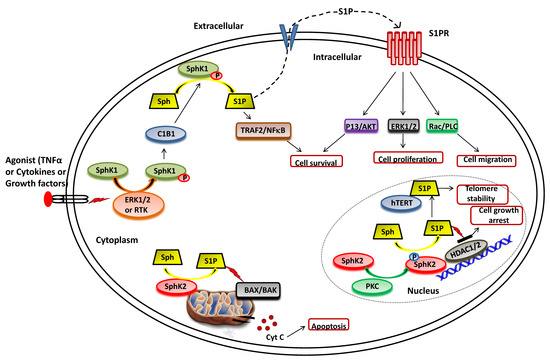
Figure 2. Functional roles of SphKs and S1P in cells. Upon ERK1/2 mediated phosphorylation/activation in the presence of various agonist (such as TNFα, cytokines and diverse growth factors), SphK1 is translocated to plasma membrane from cytoplasm and interact with calcium-myristoyl switch protein 1 (C1B1). This facilitates the phosphorylation of sphingosine to generate S1P, which can either be secreted out or interacts with intracellular targets (such as TRAF2) to elicit its functions. Once secreted out of the cell, S1P binds to the S1P receptor (S1PR) embedded in the plasma membrane and activates various downstream signaling pathways that control cell survival, proliferation, and migration. In the nucleus, SphK2 catalyzes the phosphorylation of sphingosine to generate S1P that inhibits the activity of histone deacetylases (HDAC1/2) and regulates gene expression. S1P also binds to human telomerase reverse transcriptase (hTERT) at the nuclear periphery in human and mouse fibroblasts that inhibits its interaction with makorin ring finger protein 1 (MKRN1) and promotes telomerase stability. S1P is also produced in the mitochondria by the action of SphK2.
Numerous studies suggest that the molecular mechanisms of agonist-induced stimulation involve the direct phosphorylation of SphK1 at Ser225 by ERK1/2 followed by conformational changes in the lipid kinase. This phosphorylation is crucial for the increased SphK1 activity but is required to translate the enzyme from the cytoplasm to the plasma membrane [27][35][52]. Upon activation, SphK1 interacts with calcium-myristoyl switch protein 1 that further facilitates the phosphorylation of sphingosine to S1P on the plasma membrane [53][54][55].
Several lines of evidence have consolidated the notion that SphK1 promotes cell survival. The elevated intracellular SphK1 levels appear to play an essential role in uncontrolled cell proliferation and metastasis in various cancer cell types [20][43][51][56][57][58]. A correlation between the expressions of SphK1 with short patient survival has also been observed. Brocklyn et al. [59] have demonstrated that SphK1 expression is inversely correlated with patient survival in glioblastoma multiforme. Multiple studies have illustrated that the targeted inhibition of SphK1 activity can be considered a potential strategy to combat cancer [1][43][60][61][62][63]. Likewise, the down-regulation of SphK1 via targeted inhibition induces apoptosis and enhances the sensitivity of cancer cell lines towards chemo- and radiation therapy [64][65]. Following these reports, a study showed the enhanced apoptosis in cardiomyocytes that lacks SphK1 compared to wild-type control cells under hypoxic conditions. In this case, monoganglioside (GM-1) administration improves the survival of wild-type adult mouse cardiomyocytes subjected to hypoxic and glucose deprivation conditions; however, it showed no effect on SphK1-null myocytes indicates that the activation of SphK1 by GM-1 in wild-type cells leads to cell survival [66].
2.2. SphK2 Activation and Functions
As illustrated in Figure 2, SphK2 is primarily localized to the ER or associated with mitochondria [27][33]. It also shuttles in and out of the nucleus with the assistance of nuclear localization and nuclear exportation signals possessed by the enzyme [67][68]. Earlier studies suggested apoptosis-promoting effects of SphK2 in contrast to the pro-survival action of SphK1. The overexpression of SphK2 often enhances apoptosis and cell cycle arrest [33]. The pro-apoptotic effects of this isoform have been mediated via a putative BH3 motif that interacts with the pro-survival Bcl-2 family member, Bcl-xL [69][70]. Moreover, the mitochondrial localization of SphK2 with subsequent S1P synthesis has been shown to confer the BID-mediated activation of BAK that modulates the membrane potential with the consequential release cytochrome c [29][71][72].
It has been shown that S1P produced by SphK2 in the nucleus inhibits HDAC1/2 activity that leads to enhanced histone acetylation at a specific promoter (Figure 2). This amplifies the transcription of cyclin-dependent kinase inhibitor p21 and transcriptional regulator c-fos genes, which would likely contribute to cell growth arrest attributed to SphK2 [72][73][74]. In conjunction with these findings, Weigert et al. [75] have demonstrated that knockdown of SphK2 could impede the enhanced apoptotic rates in cancer cells induced by the administration of either TNF-α or staurosporine. The genetic ablation of SphK2 suppressed the tumor growth in breast tumor xenografts (MCF-7). Contrary to the notion that SphK2 has apoptotic functions, several experimental studies have suggested an essential role of this isoenzyme in cell proliferation and survival similar to SphK1 in cancer cells [76][77][78][79]. Xu et al. [76] demonstrated that SphK2 is up-regulated in human osteosarcoma tissues and promotes cellular growth. Silencing of SphK2 by targeted shRNAs induces cell apoptosis and inhibits osteosarcoma cell growth. In another study, Xun et al. [79] showed that targeting SphK2 with a small molecule inhibitor, ABC294640, induces cell growth inhibition and apoptosis colorectal cancer cells.
Other intracellular targets of SphK2 include TERT and prohibitin. Selvam et al. [80] have shown that S1P, produced by the activity of SphK2, binds to human telomerase reverse transcriptase (hTERT) at the nuclear periphery in humans and mice fibroblasts. The binding of S1P to hTERT inhibits its interaction with makorin ring finger protein 1 (MKRN1), an E3 ubiquitin ligase that tags hTERT for degradation. Thus, S1P binding to hTERT promotes telomerase stability, telomere maintenance, cell proliferation, and tumor growth. In the mitochondria, S1P is mainly produced by SphK2 and binds with high affinity and specificity to prohibitin 2 (PHB2), a highly conserved protein that regulates cytochrome-c oxidase assembly and mitochondrial respiration [81].
3. Sphingosine-1-Phosphate and Its Receptors in the Pathophysiology
S1P acts as a second lipid messenger and regulates various physiological processes, crucial for both normal and pathological cellular conditions such as cell proliferation, migration, inflammation, and angiogenesis. S1P exerts its effects via binding to a family of five G-protein coupled receptors (GPCR), namely S1P receptor 1-5 (S1PR1-5) [1][37][82][83] (Figure 2). The autocrine or paracrine binding of S1P to S1P1-5 activates varieties of downstream signaling pathways that account for its role in many pathophysiological conditions, including cancer and inflammation [84][85][86]. Figure 3 illustrates different downstream targets of S1PRs that are vital for tumorigenesis.
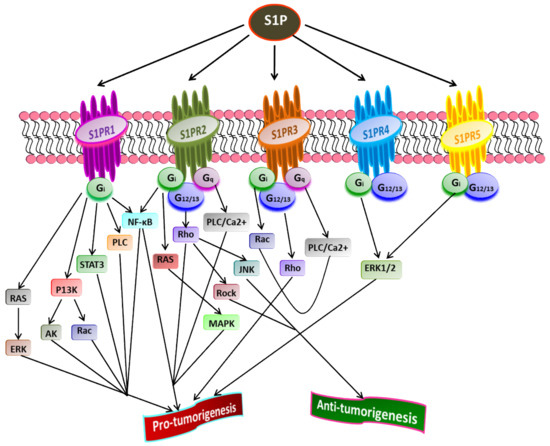
Figure 3. Role of S1P receptors in cancer. Once exported outside the cell, S1P binds and activates S1PR1-5 that further stimulates receptor-bound G-proteins (G12/13, Gq, Gi). Subsequently, the activated G-proteins turn on diverse downstream signaling pathways that play a crucial role in tumorigenesis. A cross-talk between various downstream targets of different S1PRs regulating cancer progression is evident (Adapted from [87]).
Liu et al. [88] have shown that binding S1P to S1PR1 in B cell lymphoma promotes tumor development. The inhibition of S1PR1 expression down-regulates STAT3 function leading to halting in tumor cell survival and invasion. High S1PR1 expression is associated with poor clinical outcome and the expression of several anti-apoptotic markers in non-muscle-invasive urothelial carcinoma [89]. In another study, Liu et al. [90] found the up-regulation of S1PR1 expression in bladder cancer tissues positively correlated with the density of tumor-infiltered specific regulatory T-cells in low survival rate in bladder cancer patients.
S1PR2 has been shown to promote cell proliferation and survival in acute myeloid leukemia and induce the expression of ezrin-radixin-moesin (ERM) proteins that encourage cell migration and invasion in cultured HeLa cells [91]. Recently, Kennedy et al. [92] found that S1PR2 promotes esophageal adenocarcinoma progression and cancer stem cell expansion via yes-associated protein (YAP) and β-catenin activation. Contrary to these findings, Petti et al. [93] have suggested that S1PR2 negatively correlates with the proliferation of epithelial cells in colorectal cancer. Complementing this study, Stelling et al. [94] showed the tumor-suppressive function of S1PR2 through TGFβ/TGF-β2/SMAD1 signaling axis in diffuse large B-cell lymphoma.
S1PR3 is overexpressed in breast cancer cell lines, promoting cancer progression and reducing the overall survival rate in breast cancer patients [56][95]. Hirata et al. [96] established the role of S1P/S1PR3 signaling with subsequent Notch activation in promoting tumorigenicity in aldehyde dehydrogenase-positive cancer stem cells. Moreover, they showed that tumor development in cancer stem cells could be inhibited by administering S1PR3 antagonist or S1PR3 knockdown. Recently, Shen et al. [97] found that S1PR3 is upregulated in osteosarcoma and that S1P/S1PR3 axis promotes cell proliferation and aerobic glycolysis in osteosarcoma cells. Furthermore, they showed a synergistic inhibitory action of methotrexate and S1PR3 antagonist TY52156 on osteosarcoma cellular growth.
Less is known regarding the functions of S1PR4 and S1PR5 in pathophysiology compared to other S1PRs. However, the current findings suggest that both S1PR4 and S1PR5 are coupled to Gi and G12/13 [98][99][100]. S1P/S1PR4 signaling activates downstream targets, ERK1/2, and human epidermal growth factor receptor 2 (EGFR2) pathways, thereby increasing tumorigenicity. A positive correlation was found between the overexpression of SIPR4 and shorter survival in estrogen receptor (ER)-negative breast cancer patients [95][101]. A recent study revealed the enhanced mitotic progression in HeLa cells and chromosome segregation effects through S1PR5-dependent activation of the PI3K/AKT pathway [102]. S1PR5 has been implicated in potentiating cell survival in prostate cancer cells by inducing autophagy under serum-deprived conditions [103]. In contrast, S1PR5 expression is down-regulated in esophageal squamous cell carcinoma than in normal esophageal epithelium [104]. Additionally, S1PR4 and S1PR5 play a crucial role in inflammation, often linked to the progression of some cancers, such as colon cancer [105]. Overall, the downstream signaling pathways activated by S1P through S1PR1-5play central roles in regulating the cell proliferation, survival, migration, and invasion of cancer cells [87].
Although several studies have focused on S1P signaling through S1PRs; various lines of evidence suggest the presence of direct intracellular targets of S1P. Histone deacetylases, HDAC1 and HDAC2, were among the first identified intracellular targets whose activity is inhibited by the binding of S1P, and accounts for the epigenetic regulation of specific genes inside the nucleus (Figure 3) [106][73]. Subsequent studies illustrated the binding of other intracellular proteins to the S1P, such as TNF receptor-associated factor 2 (TRAF2); an E3 ubiquitin ligase that is a key mediator the NF-κB pathway induced by inflammatory signaling molecule TNF-α [107][108]. S1P has also been shown to modulate the activity of β-amyloid precursor protein cleaving enzyme 1 implicated in Alzheimer’s disease [109]. It is now well established that S1P mediates its functions via both extracellular and intracellular signaling pathways. However, more research has to be undertaken to unravel its intracellular targets.
References
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175.
- Yamaji, T.; Hanada, K. Sphingolipid metabolism and interorganellar transport: Localization of sphingolipid enzymes and lipid transfer proteins. Traffic 2015, 16, 101–122.
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96.
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150.
- Hannun, Y.A.; Obeid, L.M. Many ceramides. J. Biol. Chem. 2011, 286, 27855–27862.
- Deevska, G.M.; Nikolova-Karakashian, M.N. The expanding role of sphingolipids in lipid droplet biogenesis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1155–1165.
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gómez-Muñoz, A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010, 9, 15.
- Saddoughi, S.A.; Ogretmen, B. Diverse functions of ceramide in cancer cell death and proliferation. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 117, pp. 37–58.
- Mullen, D.T.; Obeid, M.L. Ceramide and apoptosis: Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anti Cancer Agents Med. Chem. 2012, 12, 340–363.
- Giussani, P.; Colleoni, T.; Brioschi, L.; Bassi, R.; Hanada, K.; Tettamanti, G.; Riboni, L.; Viani, P. Ceramide traffic in C6 glioma cells: Evidence for CERT-dependent and independent transport from ER to the Golgi apparatus. Biochim. Biophys. Acta 2008, 1781, 40–51.
- Meer, G.V.; Sprong, H. Membrane lipids and vesicular traffic. Curr. Opin. Cell Biol. 2004, 16, 373–378.
- Liu, Y.-Y.; Hill, R.A.; Li, Y.-T. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 117, pp. 59–89.
- Liu, Y.-Y.; Han, T.-Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001, 15, 719–730.
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. In Sphingolipids as Signaling and Regulatory Molecules; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–23.
- Jenkins, R.W.; Canals, D.; Hannun, Y.A. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell. Signal. 2009, 21, 836–846.
- Saito, M.; Saito, M. Involvement of sphingolipids in ethanol neurotoxicity in the developing brain. Brain Sci. 2013, 3, 670–703.
- Gulbins, E. Regulation of death receptor signaling and apoptosis by ceramide. Pharmacol. Res. 2003, 47, 393–399.
- Espaillat, M.P.; Shamseddine, A.A.; Adada, M.M.; Hannun, Y.A.; Obeid, L.M. Ceramide and sphingosine-1-phosphate in cancer, two faces of the sphinx. Transl. Cancer Res. 2015, 4, 484–499.
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018.
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195.
- Lewis, A.C.; Wallington-Beddoe, C.T.; Powell, J.A.; Pitson, S.M. Targeting sphingolipid metabolism as an approach for combination therapies in haematological malignancies. Cell Death Discov. 2018, 4, 72.
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, J.S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803.
- Pralhada Rao, R.; Vaidyanathan, N.; Rengasamy, M.; Mammen Oommen, A.; Somaiya, N.; Jagannath, M.R. Sphingolipid Metabolic Pathway: An Overview of Major Roles Played in Human Diseases. J. Lipids 2013, 2013, 178910.
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 2004, 382, 527–533.
- Gariä, D.A.; De Sanctis, J.B.; Shah, J.; Dumut, D.C.; Radzioch, D. Biochemistry of very-long-chain and long-chain ceramides in cystic fibrosis and other diseases: The importance of side chain. Prog. Lipid Res. 2019, 74, 130–144.
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67.
- Hatoum, D.; Haddadi, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: Challenges for SphK as an oncotarget. Oncotarget 2017, 8, 36898.
- Wang, Z.; Min, X.; Xiao, S.-H.; Johnstone, S.; Romanow, W.; Meininger, D.; Xu, H.; Liu, J.; Dai, J.; An, S.; et al. Molecular Basis of Sphingosine Kinase 1 Substrate Recognition and Catalysis. Structure 2013, 21, 798–809.
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 2017, 8, 556.
- Melendez, A.J.; Carlos-Dias, E.; Gosink, M.; Allen, J.M.; Takacs, L. Human sphingosine kinase: Molecular cloning, functional characterization and tissue distribution. Gene 2000, 251, 19–26.
- Pyne, S.; Lee, S.C.; Long, J.; Pyne, N.J. Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell. Signal. 2009, 21, 14–21.
- Gupta, P.; Khan, F.I.; Roy, S.; Anwar, S.; Dahiya, R.; Alajmi, M.F.; Hussain, A.; Rehman, M.T.; Lai, D.; Hassan, M.I. Functional implications of pH-induced conformational changes in the Sphingosine kinase 1. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 225, 117453.
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129.
- Gupta, P.; Khan, F.I.; Ambreen, D.; Lai, D.; Alajmi, M.F.; Hussain, A.; Islam, A.; Ahmad, F.; Hassan, M.I. Investigation of guanidinium chloride-induced unfolding pathway of sphingosine kinase 1. Int. J. Biol. Macromol. 2020, 147, 177–186.
- Pitson, S.M.; Moretti, P.A.B.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. Embo J. 2003, 22, 5491–5500.
- Pulkoski-Gross, M.J.; Obeid, L.M. Molecular mechanisms of regulation of sphingosine kinase 1. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2018, 1863, 1413–1422.
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta Biomembr. 2006, 1758, 2016–2026.
- Alemany, R.; van Koppen, C.J.; Danneberg, K.; ter Braak, M.; Meyer zu Heringdorf, D. Regulation and functional roles of sphingosine kinases. Naunyn-Schmiedeberg Arch. Pharmacol. 2007, 374, 413–428.
- Maceyka, M.; Alvarez, S.E.; Milstien, S.; Spiegel, S. Activation of Sphingosine Kinase 1. In Sphingolipid Biology; Springer: Tokyo, Japan, 2006; pp. 197–206.
- Nishino, S.; Yamashita, H.; Tamori, M.; Mashimo, M.; Yamagata, K.; Nakamura, H.; Murayama, T. Translocation and activation of sphingosine kinase 1 by ceramide-1-phosphate. J. Cell. Biochem. 2019, 120, 5396–5408.
- Bao, Y.; Guo, Y.; Zhang, C.; Fan, F.; Yang, W. Sphingosine Kinase 1 and Sphingosine-1-Phosphate Signaling in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 2109.
- Zhang, X.; Cai, Y.; Zhang, W.; Chen, X. Quercetin ameliorates pulmonary fibrosis by inhibiting SphK1/S1P signaling. Biochem. Cell Biol. 2018, 96, 742–751.
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157.
- Liu, X.; Hong, Q.; Wang, Z.; Yu, Y.; Zou, X.; Xu, L. Transforming growth factor-b-sphingosine kinase 1/S1P signaling upregulates microRNA-21 to promote fibrosis in renal tubular epithelial cells. Exp. Biol. Med. 2016, 241, 265–272.
- Yamanaka, M.; Shegogue, D.; Pei, H.; Bu, S.; Bielawska, A.; Bielawski, J.; Pettus, B.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-band mediates TIMP-1 up-regulation. J. Biol. Chem. 2004, 279, 53994–54001.
- Nicholas, S.E.; Rowsey, T.G.; Priyadarsini, S.; Mandal, N.A.; Karamichos, D. Unravelling the interplay of sphingolipids and TGF-b signaling in the human corneal stroma. PLoS ONE 2017, 12, e0182390.
- Xia, P.; Wang, L.; Moretti, P.A.B.; Albanese, N.; Chai, F.; Pitson, S.M.; D’Andrea, R.J.; Gamble, J.R.; Vadas, M.A. Sphingosine Kinase Interacts with TRAF2 and Dissects Tumor Necrosis Factor-a Signaling. J. Biol. Chem. 2002, 277, 7996–8003.
- Zhang, W.; An, J.; Jawadi, H.; Siow, D.L.; Lee, J.-F.; Zhao, J.; Gartung, A.; Maddipati, K.R.; Honn, K.V.; Wattenberg, B.W.; et al. Sphingosine-1-phosphate receptor-2 mediated NFkB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat. 2013, 106, 62–71.
- Bryan, L.; Kordula, T.; Spiegel, S.; Milstien, S. Regulation and functions of sphingosine kinases in the brain. Biochim. Biophys. Acta 2008, 1781, 459–466.
- Doll, F.; Pfeilschifter, J.; Huwiler, A. Prolactin upregulates sphingosine kinase-1 expression and activity in the human breast cancer cell line MCF7 and triggers enhanced proliferation and migration. Endocr. Relat. Cancer 2007, 14, 325–335.
- Maczis, M.A.; Maceyka, M.; Waters, M.R.; Newton, J.; Singh, M.; Rigsby, M.F.; Turner, T.H.; Alzubi, M.A.; Harrell, J.C.; Milstien, S.; et al. Sphingosine kinase 1 activation by estrogen receptor a ± 36 contributes to tamoxifen resistance in breast cancer. J. Lipid Res. 2018, 59, 2297–2307.
- Pitson, S.M.; Xia, P.; Leclercq, T.M.; Moretti, P.A.B.; Zebol, J.R.; Lynn, H.E.; Wattenberg, B.W.; Vadas, M.A. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J. Exp. Med. 2005, 201, 49–54.
- Jarman, K.E.; Moretti, P.A.B.; Zebol, J.R.; Pitson, S.M. Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium-and integrin-binding protein 1. J. Biol. Chem. 2010, 285, 483–492.
- Norris, J.S. The Role of Sphingolipids in Cancer Development and Therapy; Academic Press: Cambridge, MA, USA, 2012.
- Pitson, S.M. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 2011, 36, 97–107.
- Wang, S.; Liang, Y.; Chang, W.; Hu, B.; Zhang, Y. Triple Negative Breast Cancer Depends on Sphingosine Kinase 1 (SphK1)/Sphingosine-1-Phosphate (S1P)/Sphingosine 1-Phosphate Receptor 3 (S1PR3)/Notch Signaling for Metastasis. Med. Sci. Monit. Int. Med J. Exp. Clin. Res. 2018, 24, 1912–1923.
- Wang, F.; Wu, Z. Sphingosine kinase 1 overexpression is associated with poor prognosis and oxaliplatin resistance in hepatocellular carcinoma. Exp. Ther. Med. 2018, 15, 5371–5376.
- Hart, P.C.; Chiyoda, T.; Liu, X.; Weigert, M.; Curtis, M.; Chiang, C.-Y.; Loth, R.; Lastra, R.; McGregor, S.M.; Locasale, J.W. SPHK1 is a novel target of metformin in ovarian cancer. Mol. Cancer Res. 2019, 17, 870–881.
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine Kinase-1 Expression Correlates With Poor Survival of Patients With Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705.
- Cao, M.; Ji, C.; Zhou, Y.; Huang, W.; Ni, W.; Tong, X.; Wei, J.-F. Sphingosine kinase inhibitors: A patent review. Int. J. Mol. Med. 2018, 41, 2450–2460.
- Evangelisti, C.; Evangelisti, C.; Buontempo, F.; Lonetti, A.; Orsini, E.; Chiarini, F.; Barata, J.T.; Pyne, S.; Pyne, N.J.; Martelli, A.M. Therapeutic potential of targeting sphingosine kinases and sphingosine 1-phosphate in hematological malignancies. Leukemia 2016, 30, 2142–2151.
- Gupta, P.; Mohammad, T.; Dahiya, R.; Roy, S.; Noman, O.M.A.; Alajmi, M.F.; Hussain, A.; Hassan, M.I. Evaluation of binding and inhibition mechanism of dietary phytochemicals with sphingosine kinase 1: Towards targeted anticancer therapy. Sci. Rep. 2019, 9, 18727.
- Gupta, P.; Mohammad, T.; Khan, P.; Alajmi, M.F.; Hussain, A.; Rehman, M.T.; Hassan, M.I. Evaluation of ellagic acid as an inhibitor of sphingosine kinase 1: A targeted approach towards anticancer therapy. Biomed. Pharmacother. 2019, 118, 109245.
- Haddadi, N.; Lin, Y.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. “Dicing and splicing” sphingosine kinase and relevance to cancer. Int. J. Mol. Sci. 2017, 18, 1891.
- Heffernan-Stroud, L.A.; Obeid, L.M. Sphingosine kinase 1 in cancer. Adv. Cancer Res. 2013, 117, 201–235.
- Tao, R.; Zhang, J.; Vessey, D.A.; Honbo, N.; Karliner, J.S. Deletion of the sphingosine kinase-1 gene influences cell fate during hypoxia and glucose deprivation in adult mouse cardiomyocytes. Cardiovasc. Res. 2007, 74, 56–63.
- Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S.-I. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 2003, 278, 46832–46839.
- Ding, G.; Sonoda, H.; Yu, H.; Kajimoto, T.; Goparaju, S.K.; Jahangeer, S.; Okada, T.; Nakamura, S.-I. Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J. Biol. Chem. 2007, 282, 27493–27502.
- Liu, H.; Toman, R.E.; Goparaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine Kinase Type 2 Is a Putative BH3-only Protein That Induces Apoptosis. J. Biol. Chem. 2003, 278, 40330–40336.
- Ding, X.; Zhang, Y.; Huang, T.; Xu, G.; Peng, C.; Chen, G.; Kong, B.; Friess, H.; Shen, S.; Lv, Y.; et al. Targeting sphingosine kinase 2 suppresses cell growth and synergizes with BCL2/BCL-XL inhibitors through NOXA-mediated MCL1 degradation in cholangiocarcinoma. Am. J. Cancer Res. 2019, 9, 546–561.
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000.
- Neubauer, H.A.; Pitson, S.M. Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J. 2013, 280, 5317–5336.
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257.
- Nitai Chand, H.; Jeremy, A.; Michael, M.; Graham, M.S.; Kuzhuvelil, B.H.; Tomasz, K.; Sheldon, M.; Sarah, S. Sphingosine Kinase 2 and S1P in the Nucleus Regulate Histone Acetylation by Inhibition of Histone Deacetylases. FASEB J. 2010, 24, 690–692.
- Weigert, A.; Tzieply, N.; von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol. Biol. Cell 2007, 18, 3810–3819.
- Xu, D.; Zhu, H.; Wang, C.; Zhao, W.; Liu, G.; Bao, G.; Cui, D.; Fan, J.; Wang, F.; Jin, H. SphK2 over-expression promotes osteosarcoma cell growth. Oncotarget 2017, 8, 105525.
- Mizutani, N.; Omori, Y.; Tanaka, K.; Ito, H.; Takagi, A.; Kojima, T.; Nakatochi, M.; Ogiso, H.; Kawamoto, Y.; Nakamura, M. Increased SPHK2 transcription of human colon cancer cells in serum-depleted culture: The involvement of CREB transcription factor. J. Cell. Biochem. 2015, 116, 2227–2238.
- Hasanifard, L.; Sheervalilou, R.; Majidinia, M.; Yousefi, B. New insights into the roles and regulation of SphK2 as a therapeutic target in cancer chemoresistance. J. Cell. Physiol. 2019, 234, 8162–8181.
- Xun, C.; Chen, M.-B.; Li, Q.; Zhang, T.-N.; Peng, X.; Ning, L.; Chen, Z.-H.; Wang, L.-W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94.
- Panneer Selvam, S.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58.
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612.
- Im, D.-S.; Clemens, J.; Macdonald, T.L.; Lynch, K.R. Characterization of the Human and Mouse Sphingosine 1-Phosphate Receptor, S1P5 (Edg-8): Structure− Activity Relationship of Sphingosine1-Phosphate Receptors. Biochemistry 2001, 40, 14053–14060.
- Okamoto, Y.; Wang, F.; Yoshioka, K.; Takuwa, N.; Takuwa, Y. Sphingosine-1-Phosphate-Specific G Protein-Coupled Receptors as Novel Therapeutic Targets for Atherosclerosis. Pharmaceuticals 2011, 4, 117–137.
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. 2007, 18, 300–307.
- Leong, W.I.; Saba, J.D. S1P metabolism in cancer and other pathological conditions. Biochimie 2010, 92, 716–723.
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195.
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295.
- Liu, Y.; Deng, J.; Wang, L.; Lee, H.; Armstrong, B.; Scuto, A.; Kowolik, C.; Weiss, L.M.; Forman, S.; Yu, H. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. BloodJ. Am. Soc. Hematol. 2012, 120, 1458–1465.
- Go, H.; Kim, P.-J.; Jeon, Y.K.; Cho, Y.M.; Kim, K.; Park, B.-H.; Ku, J.Y. Sphingosine-1-phosphate receptor 1 (S1PR1) expression in non-muscle invasive urothelial carcinoma: Association with poor clinical outcome and potential therapeutic target. Eur. J. Cancer 2015, 51, 1937–1945.
- Liu, Y.-N.; Zhang, H.; Zhang, L.; Cai, T.-T.; Huang, D.-J.; He, J.; Ni, H.-H.; Zhou, F.-J.; Zhang, X.-S.; Li, J. Sphingosine 1 phosphate receptor-1 (S1P1) promotes tumor-associated regulatory T cell expansion: Leading to poor survival in bladder cancer. Cell Death Dis. 2019, 10, 50.
- Orr Gandy, K.A.; Adada, M.; Canals, D.; Carroll, B.; Roddy, P.; Hannun, Y.A.; Obeid, L.M. Epidermal growth factor-induced cellular invasion requires sphingosine-1-phosphate/sphingosine-1-phosphate 2 receptor-mediated ezrin activation. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 27, 3155–3166.
- Kennedy, L.; Alpini, G. Therapeutic Role of Sphingosine-1-Phosphate Receptor 2 in the Progression of Esophageal Adenocarcinoma. Am. J. Pathol. 2018, 188, 1949–1952.
- Petti, L.; Piontini, A.; Arena, V.; Danese, S.; Vetrano, S. Sphingosine-1-phosphate receptor 2 is a negative regulator of epithelial cell proliferation and intestinal tumorigenesis. FASEB J. 2017, 31, 1046.2.
- Stelling, A.; Hashwah, H.; Bertram, K.; Manz, M.G.; Tzankov, A.; Muller, A. The tumor suppressive TGF-b/SMAD1/S1PR2 signaling axis is recurrently inactivated in diffuse large B-cell lymphoma. Blood J. Am. Soc. Hematol. 2018, 131, 2235–2246.
- Watters, R.J.; Wang, H.-G.; Sung, S.-S.; Loughran, T.P.; Liu, X. Targeting sphingosine-1-phosphate receptors in cancer. Anti-Cancer Agents Med. Chem. 2011, 11, 810–817.
- Hirata, N.; Yamada, S.; Shoda, T.; Kurihara, M.; Sekino, Y.; Kanda, Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat. Commun. 2014, 5, 1–14.
- Shen, Y.; Zhao, S.; Wang, S.; Pan, X.; Zhang, Y.; Xu, J.; Jiang, Y.; Li, H.; Zhang, Q.; Gao, J.; et al. S1P/S1PR3 axis promotes aerobic glycolysis by YAP/c-MYC/PGAM1 axis in osteosarcoma. EBioMedicine 2019, 40, 210–223.
- Siehler, S.; Manning, D.R. Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2002, 1582, 94–99.
- Turpin-Nolan, S.M.; Bruning, J.C. The role of ceramides in metabolic disorders: When size and localization matters. Nat. Rev. Endocrinol. 2020, 16, 224–233.
- Patmanathan, S.N.; Wang, W.; Yap, L.F.; Herr, D.R.; Paterson, I.C. Mechanisms of sphingosine 1-phosphate receptor signalling in cancer. Cell. Signal. 2017, 34, 66–75.
- Long, J.S.; Fujiwara, Y.; Edwards, J.; Tannahill, C.L.; Tigyi, G.; Pyne, S.; Pyne, N.J. Sphingosine 1-phosphate receptor 4 uses HER2 (ERBB2) to regulate extracellular signal regulated kinase-1/2 in MDA-MB-453 breast cancer cells. J. Biol. Chem. 2010, 285, 35957–35966.
- Andrieu, G.; Ledoux, A.; Branka, S.; Bocquet, M.; Gilhodes, J.; Walzer, T.; Kasahara, K.; Inagaki, M.; Sabbadini, R.A.; Cuvillier, O. Sphingosine 1-phosphate signaling through its receptor S1P5 promotes chromosome segregation and mitotic progression. Sci. Signal. 2017, 10, eaah4007.
- Chang, C.-L.; Ho, M.-C.; Lee, P.-H.; Hsu, C.-Y.; Huang, W.-P.; Lee, H. S1P5 is required for sphingosine 1-phosphate-induced autophagy in human prostate cancer PC-3 cells. Am. J. Physiol. Cell Physiol. 2009, 297, C451–C458.
- Hu, W.-M.; Li, L.; Jing, B.-Q.; Zhao, Y.-S.; Wang, C.-L.; Feng, L.; Xie, Y.-E. Effect of S1P5 on proliferation and migration of human esophageal cancer cells. World J. Gastroenterol. WJG 2010, 16, 1859.
- Aoki, M.; Aoki, H.; Ramanathan, R.; Hait, N.C.; Takabe, K. Sphingosine-1-phosphate signaling in immune cells and inflammation: Roles and therapeutic potential. Mediat. Inflamm. 2016, 2016.
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60.
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088.
- Park, E.-S.; Choi, S.; Shin, B.; Yu, J.; Yu, J.; Hwang, J.-M.; Yun, H.; Chung, Y.-H.; Choi, J.-S.; Choi, Y. Tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting protein (TRIP) negatively regulates the TRAF2 ubiquitin-dependent pathway by suppressing the TRAF2-sphingosine 1-phosphate (S1P) interaction. J. Biol. Chem. 2015, 290, 9660–9673.
- Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6850–6857.


