 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pouya Javadian | + 2492 word(s) | 2492 | 2021-04-22 06:18:27 | | | |
| 2 | Lindsay Dong | Meta information modification | 2492 | 2021-04-27 04:23:11 | | | | |
| 3 | Lindsay Dong | + 1 word(s) | 2493 | 2021-04-27 04:25:00 | | | | |
| 4 | Lindsay Dong | + 1 word(s) | 2493 | 2021-04-27 04:25:29 | | | | |
| 5 | Lindsay Dong | Meta information modification | 2493 | 2021-04-27 04:26:31 | | | | |
| 6 | Lindsay Dong | Meta information modification | 2493 | 2021-04-27 04:29:19 | | | | |
| 7 | Conner Chen | Meta information modification | 2493 | 2021-09-22 02:53:13 | | |
Video Upload Options
In contrast to the decline in incidence and mortality of most other cancers, these rates are rising for endometrial cancer. Black women with endometrial cancer have an earlier diagnosis, more aggressive histology, advanced stage, and worse outcomes compared with their White counterparts. Socioeconomic status, a higher incidence of aggressive histology, and comorbid conditions are known factors leading to racial disparity in patients with endometrial cancer; nevertheless, they do not account for the entire racial disparity, which emphasizes the roles of molecular, histopathological and genetic factors.
1. Introduction
Black patients are diagnosed and die earlier of endometrial cancer in comparison with their White counterparts. Socioeconomic status, a higher incidence of aggressive histology, and comorbid conditions are known factors leading to racial disparity in patients with endometrial cancer; nevertheless, they do not account for the entire racial disparity; which emphasizes the roles of molecular, histopathological and genetic factors. Although the incidence of endometrial cancer has been reduced by 30% among Black patients, the mortality of endometrial cancer is 2.5 times higher in comparison with White patients [1]. This represents one of the largest racial disparities in mortality among common cancers [2]. The incidence of uterine cancers has continued to rise over time, while poorer survival continues to be reported among Black patients [3], even after adjusting for age and hysterectomy [4].
The contributions of socioeconomic factors to endometrial cancer racial disparities have been reviewed elsewhere [5][6]. In this study, we investigate the current body of literature to identify molecular, biological, histopathologic, and genetic factors associated with endometrial cancer disparities and identify potential pharmaceutical interventions.
2. Endometrial Cancer Racial Disparity
2.1. Histology, Pathogenic Types and Endometrial Disparity
Endometrial cancers are categorized into different histologic subtypes including: endometrioid (75–80% of patients), USC (<10% of patients), clear cell (<5% of patients) and carcinosarcoma (<5% of patients) [3]. Type I tumors comprise the large majority of endometrial cancers, develop from and are associated with atypical glandular hyperplasia, are related to unopposed estrogen stimulation and are often preceded by endometrial hyperplasia. Type II histological subtypes include predominantly USC, and also clear cell, carcinosarcoma, high-grade endometroid cancer, and mixed (typically endometrioid and a high-grade nonendometrioid pattern) [7]. In multiple studies, Black endometrial cancer patients had higher incidences of these aggressive histologic subtypes in comparison with non-Hispanic White patients [8][9][10][11][12][13][14]. Overall, Black patients have the highest incidence of these most aggressive, nonendometrioid histologies of endometrial cancer in the US population (Figure 1) [10].
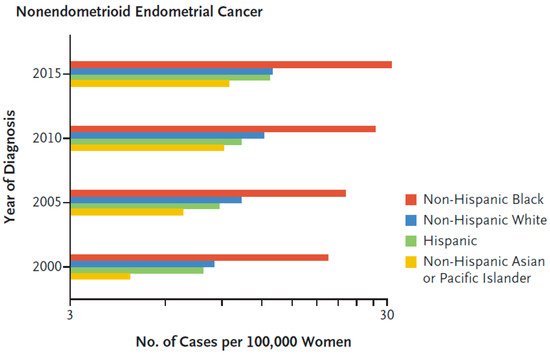
Figure 1. Age-adjusted and hysterectomy-corrected incidence of nonendometrioid endometrial cancer according to race [10]. Reproduced with permission from Massachusetts Medical Society.
The increased incidence of Type II compared with Type I tumors is consistent with the worse 5-year disease-specific survival of Black (51–57%), compared with White (65–67%), Hispanic (64–69%) and Asian (67–72%) endometrial cancer patients in a California registry study (p < 0.0001) [8][15]. In a Florida registry study, Black endometrial cancer patients had a higher incidence of Type II tumors (57.6%) compared with White (35.6%), Hispanic (37.7%), Asian (43.0%) endometrial cancer patients, and a 24% high risk of death due to endometrial cancer compared with white patients [15]. A national population-based study of 35,850 young women (<50 years of age) with endometrial cancer found that the young Black patients (3903) had higher mortality compared with the young White patients [14].
2.2. Hereditary Cancer and Genetic Predisposition
A study of a sample population of 62 Black and 78 White patients diagnosed with Stage III/IV endometrial adenocarcinomas found no racial disparities in the frequency of MSI (Whites, 16%; Blacks, 13%), in the subset with endometrioid histology (Whites, 20%; Blacks, 22%) [16]. Analysis of the TCGA dataset revealed that patients of Asian descent have a higher frequency of somatic mutations of MMR genes in contrast with White or Black groups of patients. Missense mutation is the most prevalent type of mutation in the Asian population and the MMR-associated gene, PMS2, was identified as the most significant mutated gene among all other genes in the TCGA dataset (Fisher’s exact test; p = 0.0036) [17].
2.3. Global Profiling to Identify Racial Differences in mRNA Transcripts and Proteins
Publications of global profiling efforts to identify mRNA and proteins that could contribute to racial disparities in endometrial cancer are limited to two studies comparing endometrial cancer specimens in Black and White patients. The molecules identified in these studies are summarized in Table 1. The proteomic analysis identified 94 proteins as being altered between the Black and White groups. Ten of these candidates were reduced in expression at both the RNA and protein levels in cancers from Black compared with White patients (Table 1) [18].
Table 1. Molecules significantly associated with race and survival.
| Difference | Name (Gene Symbol) | Survival Association | Reference |
|---|---|---|---|
| RNA Increased in Black Patients | DSN1 homolog (DSN1) a | PFS in Black and White | [18] |
| PBX homeobox 1 (PBX1) a,d | PFS in Black b | [18] | |
| EPM2A laforin glucan phosphatase (EPM2A)a | PFS and OS in White b | [18] | |
| DNA Ligase 3 (LIG3) a | PFS worse for Black and improved for White | [18] | |
| Bet1 Golgi vesicular membrane trafficking protein like (BET1L) a | PFS in White | [18] | |
| HEAT repeat containing 6 (HEATR6) a | PFS in Black | [18] | |
| Family with sequence similarity 228 member B (FAM228B) a | PFS in Black b | [18] | |
| Adaptor-related protein complex 4 r1 subunit (ARCA5) a | PFS in Black | [18] | |
| PSMD5 antisense RNA 1 (head to head) (PSMD5-AS1) a | PFS in Black b | [18] | |
| Polyribonucleotide nucleotidyltransferase 1 (PNPT1) a | PFS in Black | [18] | |
| Adaptor-related protein complex 4 r1 subunit (AP4S1) a | PFS in Black b | [18] | |
| Cyclin E1 (CCNE1) a,c,d | Not determined | [19] | |
| Cyclin dependent kinase inhibitor 2A/P16 (CDKN2A) c,d | Not determined | [19] | |
| Cell division cycle 25C (CDC25C) c | Not determined | [19] | |
| Cyclin B2 (CCNB2) c | Not determined | [19] | |
| Mitotic checkpoint serine/threonine kinase B (BUB1B) c | Not determined | [19] | |
| Minichromosome Maintenance complex component 7 (MCM7) c | Not determined | [19] | |
| Polo like kinase 1 (PLK1) c,d | Not determined | [19] | |
| Minichromosome maintenance complex component 2 (MCM2) c | Not determined | [19] | |
| Baculoviral inhibitor of apoptosis repeat containing 7 (BIRC7) c | Not determined | [19] | |
| Laminin subunit gamma 1 (LAMC1) c | Not determined | [19] | |
| CDC28 protein kinase regulatory subunit 1B (CKS1B) c | Not determined | [19] | |
| Laminin subunit 5 (LAMA5) c | Not determined | [19] | |
| Wnt family member 7A (WNT7A) c | Not determined | [19] | |
| Fibroblast growth factor 12 (FGF12) c | Not determined | [19] | |
| Fibroblast growth factor 5 (FGF5) c | Not determined | [19] | |
| FGF receptor 3 (FGFR3) c,d | Not determined | [19] | |
| Erb-B2 receptor tyrosine kinase 2 (ERBB2) c,d | Not determined | [19] | |
| Breast cancer 2 DNA repair associated (BRCA2) c | Not determined | [19] | |
| Phosphatidylinositol-4, 5-bisphosphate3-kinase catalytic subunit α (PIK3CA) c,d | Not determined | [19] | |
| RNA Decreased in Black Race | Wilms tumor 1 associated protein (WTAP) a | PFS in White | [18] |
| Death-associated protein (DAP) a | PFS and OS in White b | [18] | |
| Mal T-cell differentiation protein like (MALL) a | PFS in White b | [18] | |
| Integrin subunit a3 (ITGA3) a | PFS and OS in White | [18] | |
| Uncoupling protein 2 (UCP2) a | PFS in White b | [18] | |
| Serpin family A member 4 (SERPINA4) a | PFS and OS in White b | [18] | |
| Calpain 6 (CAPN6) a | PFS in White b | [18] | |
| Solute carrier family 24 member 1 (SLC24A1) a | PFS in Black | [18] | |
| Chromosome 19 open reading frame 25 (C19orf25) a | PFS in Black | [18] | |
| WD repeat domain 97 (WDR97) a | PFS in Black | [19] | |
| Cyclin A2 (CCNA2) c | Not determined | [19] | |
| Mitotic arrest deficient 2 like 1 (MAD2L1) c | Not determined | [19] | |
| Cyclin E1 (CCNE2) c | Not determined | [19] | |
| Cell division cycle 25C (CDC25A) c | Not determined | [19] | |
| Cell division cycle 7 (CDC7) c,d | Not determined | [19] | |
| Structural maintenance of chromosomes 1B (SMC1B) c | Not determined | [19] | |
| RNA and Protein Decreased in Blacks | CCR4-NOT transcription complex subunit 1 (CNOT1) | Not determined | [18] |
| Peptidylprolyl isomerase B (PPIB) | Not determined | [18] | |
| Cell cycle and apoptosis regulator 2 (CCAR2) | Not determined | [18] | |
| Eukaryotic translation elongation factor 2 (EEF2)d | Not determined | [18] | |
| Junction plakoglobin (JUP) | Not determined | [18] | |
| PDZ and LIM domain 5 (PDLIM5) | Not determined | [18] | |
| Myosin IC (MYO1C) | Not determined | [18] | |
| Desmoglein 2 (DSG2) | Not determined | [18] | |
| Plakophilin 3 (PKP3) | Not determined | [18] | |
| DnaJ heat shock protein family (Hsp40) member C10 (DNAJC10) | Not determined | [18] |
a Validated in TCGA RPPA data. b Significant in multivariate COX analysis for stage, grade and myometrial invasion; Wald p < 0.05. c Mitotic subtype only. d Actionable Target: PBX1—camptothecin [20][21]; EEF2—MDNA55 [22]; CCNE1—CDK2 and Akt inhibitors [23][24]; CDKN2A—senolytics, PLK1- BI 2536, GSK461364, lipid-encapsulated anti-PLK1 siRNA TKM-080301, MK1496, onvansertib, rigosertib, TAK-960 and volasertib [25][26]; CDC7—BMS-863233, LY3143921, NMS-1116354, TAK-93 [27][28][29][30]; FGFR3—3D185, ASP5878, B701, crizotinib/pazopanib and debio [31][32]; ERBB2—68GaNOTA-anti-HER2 VHH1, 89Zr-trastuzumab, 99m-Tc-NM-02, 99mTc-HPArk2, [68Ga] ABY-025, A166, ADCT-502, afatinib, afatinib/cetuximab, afatinib/osimertinib, afatinib/paclitaxel, allitinib and alpelisib/trastuzumab [33]; PIK3CA—alpelisib, alpelisib/fulvestrant, alpelisib/trastuzumab emtansine, AZD8835, BAY1082439, bimiralisib, buparlisib, CLR457, copanlisib, dactolisib, GDC-0077, pictilisib, pilaralisib, PKI-179, PWT33597, PX-866, serabelisib, SF 1126, taselisib [27][28][29][30]; PLK1—volasterib, fostamatinib, cytarabine, decitabine [34][35][36][37][38][39][40].
2.4. Racial Differences in MicroRNAs
A comparison of laser micro-dissected unmatched Stage I endometrioid endometrial cancer specimens from 41 White and 9 Black patients found no global differences; however, a subset analysis of stage- and histology-matched specimens from nine White and nine Black patients identified alterations in 18 microRNAs (miRNAs) [41]. Altered DNA methylation, along with increased microRNA expression levels in African-American patients, is associated with cancer drug resistance in this patient population [42].
2.5. A Targeted Approach to Identifying Racial Differences in Specific Proteins
Another approach to identifying the biological factors responsible for racial disparities are targeted studies that evaluate specific molecular candidates for differential expression and associations with survival between racial groups (Table 1). In Black patients with endometrial cancer, a higher frequency of p53 overexpression increased the chance of Type II cancer in this patient population [43]. The rate of p53 overexpression in patients with Type I cancer was not different between the two ethnic groups [43]. The rate of p53 overexpression observed in endometrial cancers from White patients was similar to the rate observed in Japanese patients reported by the same research group [44][45].
2.6. Pathway Analysis of Racial Differences in mRNA and Proteins
We conducted an Ingenuity analysis to identify associations between proteins exhibiting altered expression in endometrial cancer specimens from Black compared with White patients and that were also prognostically significant in Black patients (Figure 3). The analysis demonstrated that p53 transcription factor increases the expression of PTEN, HER2/NEU and IGF2, proteins which also are known to interact with each other. The missense p53 mutations that can cause p53 protein overexpression, however, often alter the profiles of the genes regulated by p53. HER2/NEU is an indirect upstream regulator of IGF2, LIG3 and PBX1, and IGF2 regulates PTEN expression.
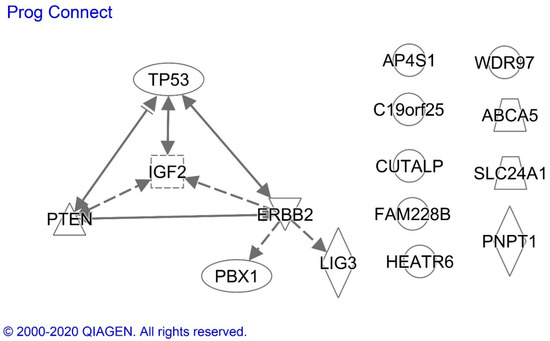
Figure 3. Ingenuity analysis of prognostically significant proteins with altered expression in endometrioid endometrial cancer specimens from Black compared with White patients. Solid and dotted lines indicate the direct and indirect effects, respectively, of the indicated protein on the protein to which the arrowhead is pointing. ERBB2 = HER2/NEU. Proteins without arrows had no evidence of a connection with the other proteins in the current scientific literature.
Analysis of all of the molecules listed in Table 1 confirmed the significant association of molecules differentially expressed in Blacks with the cell cycle regulation pathway and showed pathway associations among the cell cycle regulatory proteins along with EEF2, HER2/NEU, FGFR3 MCM2/7 and BRCA2 (Figure 4).
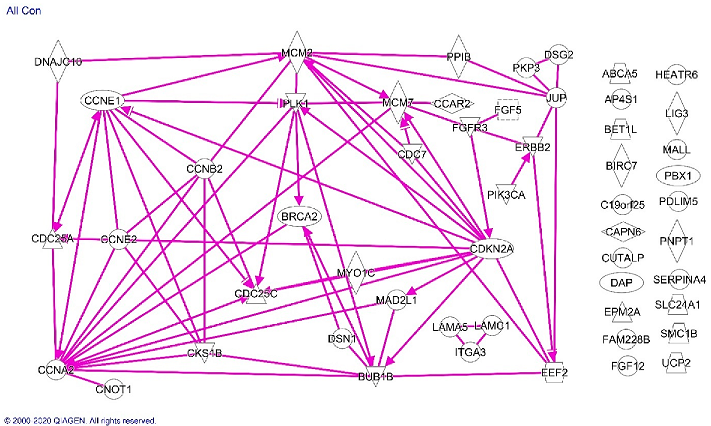
Figure 4. Ingenuity analysis of proteins differentially expressed in endometrioid endometrial cancer specimens from Black compared with White patients. Solid lines indicate direct effects of the indicated protein on the protein to which the arrowhead is pointing. Proteins without arrows had no evidence of a connection with the other proteins in the current scientific literature.
2.7. Race-Based Treatment and Outcome
A growing body of evidence suggests that targeted therapies have an improved capacity for improving cancer patient outcomes with less toxicity compared with chemotherapy [46][47]. A gynecologic oncology group study of patients receiving treatment for advanced endometrial cancer in clinical trials found that 169 Black patients experienced 20% worse survival compared with 982 White patients, even though the participants received uniform care [48]. The disparity remained when they controlled for socioeconomic status and tumor stage, grade, and histology. Further investigation demonstrated that Black patients experienced lesser responses to chemotherapy regimens consistently across several clinical studies. Taken together, these findings suggest that there are potential molecular and biological components contributing to the observed racial disparity, and thus emphasizing the promise of new molecularly-targeted therapies in Black women.
2.8. Candidate Drugs for Race-Specific Treatment of Black Endometrial Cancer Patients
Of the race-specific proteins listed in Table 2, p53 and HER2/NEU (ERBB2) have molecularly targeted agents in development or approved for cancer treatment. The higher expression of p53 protein in endometrial cancer specimens from Black patients may be due to the higher TP53 gene mutation rate associated with USC, which occurs at a higher rate compared to other histologies in Black women. There are multiple drugs targeted at HER2/NEU. A randomized phase 2 trial adding trastuzumab to chemotherapy showed promise in treating USCs that overexpress HER2/NEU [.]. Currently, there are two clinical trials of HER2/NEU inhibitors for endometrial cancer listed on clinicaltrials.gov.
Table 2. Race-Specific Proteins as Drug Targets in Endometrial Cancer.
|
Protein |
Racial Difference |
Result |
|
p53 |
Increased in Blacks |
Worse survival in all patients. |
|
PTEN |
Decreased in Blacks |
Favorable outcome. |
|
HER2/Neu |
Increased in Blacks |
Worse disease-related survival in all races |
|
IGF2 |
Increased in Blacks |
Worse survival in all races and doubling of risk in Blacks with USC. |
There are multiple drugs targeting cyclins and cyclin-dependent kinases (CDKs) in clinical trials that provide the potential for inhibiting cyclin E overexpression in endometrial cancers of Black patients[49][50].
Other molecules listed in Table 1 can be inhibited indirectly by known drugs. The expression of PBX1 is decreased by camptothecin, which is a topoisomerase poison used to treat leukemia. Multiple formulations of camptothecin analogs are currently in clinical trials for a variety of cancers (clinicaltrials.gov (access date: 10 January 2021)). EEF2 can be inactivated by a biologic drug called MDNA55, which is a circularly permuted version of interleukin-4 (IL-4) conjugated to Pseudomonas Exotoxin A (PE). Upon binding to the IL-4 receptor, the drug is internalized in the cell, where the PE is released and causes ADP-ribosylation of EEF2 on ribosomes [22]. There are also other biologics and immunotoxins that are conjugated with PE and provide additional candidate drugs for the race-specific treatment of endometrial cancer [51].
The mutational profiles of tumors have also been shown to affect sensitivities to specific cancer therapies. For example, in Phase 2 clinical trial of 86 patients with MMR deficiencies across different solid tumors, including endometrial cancer, pembrolizumab inhibition of PD-1 resulted in a 77% disease control rate [52]. In this study, genomic signatures predicted tumor phenotype and treatment responses independently of the anatomic site of the disease or the histology of the tumor. Therefore, PD1 blockade may provide improved outcomes in endometrial cancer patients of Asian descent, based on the higher frequency of somatic mutations of MMR genes in Asian endometrial cancer patients.
3. Discussion
Our review of the literature identified molecules and molecular pathways that are present at significantly different levels in endometrial cancer specimens from Black and White patients, and encourages controlled studies of other races and ethnicities. Several of these molecules and their pathways can be targeted by existing pharmaceuticals, which have the potential for use in race-specific treatment strategies. Recent studies have revealed the potential for improved patient management by enabling tumors to be classified into molecularly categorized subgroups, in addition to the four TCGA groups, with significantly different tumor responses to treatment patient prognoses. These results encourage further elucidation of molecular pathways and genetic characteristics of endometrial cancers for all race and ethnic groups exhibiting disparities. This information can be used to develop better predictive models of cancer progression and novel therapeutic approaches.
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300.
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030.
- Surveillance, E. End Results (SEER) Program. Available online: (accessed on 10 January 2021).
- Doll, K.M.; Winn, A.N. Assessing endometrial cancer risk among US women: Long-term trends using hysterectomy-adjusted analysis. Am. J. Obstet. Gynecol. 2019, 221, 318.e1–318.e9.
- Donkers, H.; Bekkers, R.; Massuger, L.; Galaal, K. Systematic review on socioeconomic deprivation and survival in endometrial cancer. Cancer Causes Control CCC 2019, 30, 1013–1022.
- Donkers, H.; Bekkers, R.; Massuger, L.; Galaal, K. Socioeconomic deprivation and survival in endometrial cancer: The effect of BMI. Gynecol. Oncol. 2020, 156, 178–184.
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; McCann, S.E.; Yu, H.; Xiang, Y.B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II endometrial cancers: Have they different risk factors? J. Clin. Oncol. 2013, 31, 2607–2618.
- Baskovic, M.; Lichtensztajn, D.Y.; Nguyen, T.; Karam, A.; English, D.P. Racial disparities in outcomes for high-grade uterine cancer: A California cancer registry study. Cancer Med. 2018, 7, 4485–4495.
- Ward, E.M.; Sherman, R.L.; Henley, S.J.; Jemal, A.; Siegel, D.A.; Feuer, E.J.; Firth, A.U.; Kohler, B.A.; Scott, S.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, Featuring Cancer in Men and Women Age 20–49 Years. J. Natl. Cancer Inst. 2019, 111, 1279–1297.
- Lu, K.H.; Broaddus, R.R. Endometrial Cancer. N. Engl. J. Med. 2020, 383, 2053–2064.
- Sherman, M.E.; Devesa, S.S. Analysis of racial differences in incidence, survival, and mortality for malignant tumors of the uterine corpus. Cancer 2003, 98, 176–186.
- Huang, A.B.; Huang, Y.; Hur, C.; Tergas, A.I.; Khoury-Collado, F.; Melamed, A.; St Clair, C.M.; Hou, J.Y.; Ananth, C.V.; Neugut, A.I.; et al. Impact of quality of care on racial disparities in survival for endometrial cancer. Am. J. Obstet. Gynecol. 2020, 223, 396.e1–396.e13.
- Santin, A.D.; Bellone, S.; Siegel, E.R.; Palmieri, M.; Thomas, M.; Cannon, M.J.; Kay, H.H.; Roman, J.J.; Burnett, A.; Pecorelli, S. Racial differences in the overexpression of epidermal growth factor type II receptor (HER2/neu): A major prognostic indicator in uterine serous papillary cancer. Am. J. Obstet. Gynecol. 2005, 192, 813–818.
- Mukerji, B.; Baptiste, C.; Chen, L.; Tergas, A.I.; Hou, J.Y.; Ananth, C.V.; Neugut, A.I.; Hershman, D.L.; Wright, J.D. Racial disparities in young women with endometrial cancer. Gynecol. Oncol. 2018, 148, 527–534.
- Johnson, A.L.; Medina, H.N.; Schlumbrecht, M.P.; Reis, I.; Kobetz, E.N.; Pinheiro, P.S. The role of histology on endometrial cancer survival disparities in diverse Florida. PLoS ONE 2020, 15, e0236402.
- Maxwell, G.L.; Risinger, J.I.; Hayes, K.A.; Alvarez, A.A.; Dodge, R.K.; Barrett, J.C.; Berchuck, A. Racial disparity in the frequency of PTEN mutations, but not microsattelite instability, in advanced endometrial cancers. Clin. Cancer Res. 2000, 6, 2999–3005.
- Guttery, D.S.; Blighe, K.; Polymeros, K.; Symonds, R.P.; Macip, S.; Moss, E.L. Racial differences in endometrial cancer molecular portraits in The Cancer Genome Atlas. Oncotarget 2018, 9, 17093–17103.
- Bateman, N.W.; Dubil, E.A.; Wang, G.; Hood, B.L.; Oliver, J.M.; Litzi, T.A.; Gist, G.D.; Mitchell, D.A.; Blanton, B.; Phippen, N.T.; et al. Race-specific molecular alterations correlate with differential outcomes for black and white endometrioid endometrial cancer patients. Cancer 2017, 123, 4004–4012.
- Dubil, E.A.; Tian, C.; Wang, G.; Tarney, C.M.; Bateman, N.W.; Levine, D.A.; Conrads, T.P.; Hamilton, C.A.; Maxwell, G.L.; Darcy, K.M. Racial disparities in molecular subtypes of endometrial cancer. Gynecol. Oncol. 2018, 149, 106–116.
- Carson, J.P.; Zhang, N.; Frampton, G.M.; Gerry, N.P.; Lenburg, M.E.; Christman, M.F. Pharmacogenomic identification of targets for adjuvant therapy with the topoisomerase poison camptothecin. Cancer Res. 2004, 64, 2096–2104.
- Brachat, A.; Pierrat, B.; Xynos, A.; Brecht, K.; Simonen, M.; Brüngger, A.; Heim, J. A microarray-based, integrated approach to identify novel regulators of cancer drug response and apoptosis. Oncogene 2002, 21, 8361–8371.
- Iglewski, B.H.; Liu, P.V.; Kabat, D. Mechanism of action of Pseudomonas aeruginosa exotoxin Aiadenosine diphosphate-ribosylation of mammalian elongation factor 2 in vitro and in vivo. Infect. Immun. 1977, 15, 138–144.
- Caruso, J.A.; Duong, M.T.; Carey, J.P.W.; Hunt, K.K.; Keyomarsi, K. Low-Molecular-Weight Cyclin E in Human Cancer: Cellular Consequences and Opportunities for Targeted Therapies. Cancer Res. 2018, 78, 5481–5491.
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488.
- Malavolta, M.; Bracci, M.; Santarelli, L.; Sayeed, M.A.; Pierpaoli, E.; Giacconi, R.; Costarelli, L.; Piacenza, F.; Basso, A.; Cardelli, M.; et al. Inducers of Senescence, Toxic Compounds, and Senolytics: The Multiple Faces of Nrf2-Activating Phytochemicals in Cancer Adjuvant Therapy. Mediat. Inflamm. 2018, 2018, 4159013.
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536.
- Drullinsky, P.R.; Hurvitz, S.A. Mechanistic basis for PI3K inhibitor antitumor activity and adverse reactions in advanced breast cancer. Breast Cancer Res. Treat. 2020, 181, 233–248.
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2019, 30, 1051–1060.
- Verret, B.; Cortes, J.; Bachelot, T.; Andre, F.; Arnedos, M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, x12–x20.
- Brandao, M.; Caparica, R.; Eiger, D.; de Azambuja, E. Biomarkers of response and resistance to PI3K inhibitors in estrogen receptor-positive breast cancer patients and combination therapies involving PI3K inhibitors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, x27–x42.
- Shen, G.; Zheng, F.; Ren, D.; Du, F.; Dong, Q.; Wang, Z.; Zhao, F.; Ahmad, R.; Zhao, J. Anlotinib: A novel multi-targeting tyrosine kinase inhibitor in clinical development. J. Hematol. Oncol. 2018, 11, 120.
- Roubal, K.; Myint, Z.W.; Kolesar, J.M. Erdafitinib: A novel therapy for FGFR-mutated urothelial cancer. Am. J. Health-Syst. Pharm. 2020, 77, 346–351.
- Akbari, V.; Chou, C.P.; Abedi, D. New insights into affinity proteins for HER2-targeted therapy: Beyond trastuzumab. Biochim. Et Biophys. Acta Rev. Cancer 2020, 1874, 188448.
- Van den Bossche, J.; Lardon, F.; Deschoolmeester, V.; De Pauw, I.; Vermorken, J.B.; Specenier, P.; Pauwels, P.; Peeters, M.; Wouters, A. Spotlight on Volasertib: Preclinical and Clinical Evaluation of a Promising Plk1 Inhibitor. Med. Res. Rev. 2016, 36, 749–786.
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Rentschler, J.; Kaiser, R.; Rouyrre, N.; Trommeshauser, D.; Hoesl, C.E.; Munzert, G. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5511–5517.
- Schoffski, P.; Blay, J.-Y.; De Greve, J.; Brain, E.; Machiels, J.-P.; Soria, J.-C.; Sleijfer, S.; Wolter, P.; Ray-Coquard, I.; Fontaine, C.; et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network of Core Institutes (NOCI). Eur. J. Cancer 2010, 46, 2206–2215.
- Hofheinz, R.-D.; Al-Batran, S.-E.; Hochhaus, A.; Jager, E.; Reichardt, V.L.; Fritsch, H.; Trommeshauser, D.; Munzert, G. An open-label, phase I study of the polo-like kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 4666–4674.
- Olmos, D.; Barker, D.; Sharma, R.; Brunetto, A.T.; Yap, T.A.; Taegtmeyer, A.B.; Barriuso, J.; Medani, H.; Degenhardt, Y.Y.; Allred, A.J.; et al. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 3420–3430.
- Breitenbuecher, F.; von Pawel, J.; Sebastian, M.; Kortsik, C.; Ting, S.; Kasper, S.; Wohlschlager, J.; Worm, K.; Morresi-Hauf, A.; Schad, A.; et al. Comprehensive Biomarker Analyses in Patients with Advanced or Metastatic Non-Small Cell Lung Cancer Prospectively Treated with the Polo-Like Kinase 1 Inhibitor BI2536. Oncol. Res. Treat. 2017, 40, 435–439.
- Weiss, G.J.; Jameson, G.; Von Hoff, D.D.; Valsasina, B.; Davite, C.; Di Giulio, C.; Fiorentini, F.; Alzani, R.; Carpinelli, P.; Di Sanzo, A.; et al. Phase I dose escalation study of NMS-1286937, an orally available Polo-Like Kinase 1 inhibitor, in patients with advanced or metastatic solid tumors. Investig. New Drugs 2018, 36, 85–95.
- Maxwell, G.L.; Shoji, Y.; Darcy, K.; Litzi, T.; Berchuck, A.; Hamilton, C.A.; Conrads, T.P.; Risinger, J.I. MicroRNAs in endometrial cancers from black and white patients. Am. J. Obs. Gynecol 2015, 212, 191.e1–191.e10.
- Lara, O.D.; Wang, Y.; Asare, A.; Xu, T.; Chiu, H.S.; Liu, Y.; Hu, W.; Sumazin, P.; Uppal, S.; Zhang, L.; et al. Pan-cancer clinical and molecular analysis of racial disparities. Cancer 2020, 126, 800–807.
- Schimp, V.L.; Ali-Fehmi, R.; Solomon, L.A.; Hammoud, A.; Pansare, V.; Morris, R.T.; Munkarah, A.R. The racial disparity in outcomes in endometrial cancer: Could this be explained on a molecular level? Gynecol. Oncol. 2006, 102, 440–446.
- Inoue, M.; Okayama, A.; Fujita, M.; Enomoto, T.; Sakata, M.; Tanizawa, O.; Ueshima, H. Clinicopathological characteristics of p53 overexpression in endometrial cancers. Int. J. Cancer 1994, 58, 14–19.
- Ito, K.; Watanabe, K.; Nasim, S.; Sasano, H.; Sato, S.; Yajima, A.; Silverberg, S.G.; Garrett, C.T. Prognostic significance of p53 overexpression in endometrial cancer. Cancer Res. 1994, 54, 4667–4670.
- Ott, P.A.; Bang, Y.J.; Berton-Rigaud, D.; Elez, E.; Pishvaian, M.J.; Rugo, H.S.; Puzanov, I.; Mehnert, J.M.; Aung, K.L.; Lopez, J.; et al. Safety and Antitumor Activity of Pembrolizumab in Advanced Programmed Death Ligand 1-Positive Endometrial Cancer: Results From the KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 2535–2541.
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196.
- Maxwell, G.L.; Tian, C.; Risinger, J.; Brown, C.L.; Rose, G.S.; Thigpen, J.T.; Fleming, G.F.; Gallion, H.H.; Brewster, W.R.; Gynecologic Oncology Group, s. Racial disparity in survival among patients with advanced/recurrent endometrial adenocarcinoma: A Gynecologic Oncology Group study. Cancer 2006, 107, 2197–2205.
- Joseph A. Caruso; Mylinh T. Duong; Jason P. W. Carey; Kelly K. Hunt; Khandan Keyomarsi; Low-Molecular-Weight Cyclin E in Human Cancer: Cellular Consequences and Opportunities for Targeted Therapies. Cancer Research 2018, 78, 5481-5491, 10.1158/0008-5472.can-18-1235.
- Robert Roskoski; Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacological Research 2018, 139, 471-488, 10.1016/j.phrs.2018.11.035.
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219.
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413.

