 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Luisa Brandi | + 1416 word(s) | 1416 | 2021-04-23 08:21:22 | | | |
| 2 | Nora Tang | Meta information modification | 1416 | 2021-04-25 08:41:19 | | |
Video Upload Options
Pancreatic neuroendocrine tumors (pNETs) are a rare group of cancers accounting for about 1–2% of all pancreatic neoplasms. About 10% of pNETs arise within endocrine tumor syndromes, such as Multiple Endocrine Neoplasia type 1 (MEN1). pNETs affect 30–80% of MEN1 patients, manifesting prevalently as multiple microadenomas. pNETs in patients with MEN1 are particularly difficult to treat due to differences in their growth potential, their multiplicity, the frequent requirement of extensive surgery, the high rate of post-operative recurrences, and the concomitant development of other tumors. MEN1 syndrome is caused by germinal heterozygote inactivating mutation of the MEN1 gene, encoding the menin tumor suppressor protein. MEN1-related pNETs develop following the complete loss of function of wild-type menin. Menin is a key regulator of endocrine cell plasticity and its loss in these cells is sufficient for tumor initiation. Somatic biallelic loss of wild-type menin in the neuroendocrine pancreas presumably alters the epigenetic control of gene expression, mediated by histone modifications and DNA hypermethylation, as a driver of MEN1-associated pNET tumorigenesis. In this light, epigenetic-based therapies aimed to correct the altered DNA methylation, and/or histone modifications might be a possible therapeutic strategy for MEN1 pNETs, for whom standard treatments fail.
1. Classification
Pancreatic neuroendocrine tumors (pNETs) are rare primary cancers of the endocrine pancreas accounting less than 3% of all pancreatic neoplasms. pNETs manifest mainly as sporadic tumors, or arise in the context of genetically determined syndromes in less than 10% of cases.
pNETs are primarily distinguished between functioning (15%) and nonfunctioning tumors (85%). Functioning pNETs retain secretory ability and release excessive amounts of hormones (i.e., insulin, gastrin, glucagon), which cause specific endocrine syndromes and related symptoms. Conversely, nonfunctioning pNETs (NF-pNETs) are more poorly differentiated tumors which do not secrete any hormone or active peptide, are often asymptomatic, and are discovered incidentally through abdominal screening performed for other reasons, or if their growth causes a symptom-inducing compression of adjacent structures (i.e., obstruction of the pancreas/bile duct), usually when they have already metastasized the adjacent lymph nodes and/or the liver. NF-pNETs have a worse prognosis; patients present a high percentage of unresectable disease, often with liver metastasis, and a five-year survival rate of 30–40% [1].
According to the 2017 WHO classification for pNETs [2] and the 2019 WHO classification of tumors of the digestive system [3], pNETs and pancreatic neuroendocrine carcinomas (pNECs) are included in the superfamily of pancreatic neuroendocrine neoplasms (pNENs).
2. Pathology
Pathological classification of all pNENs includes tumor grade and malignity; pNENs can be ascribed three different grades based on tumor cell proliferation rate, determined by mitotic count and/or Ki-67 nuclear staining index [2]. Grade 1 (G1) includes well-differentiated low-grade pNETs with a Ki-67 index less than 3% of ≥500 cells and/or a mitotic count less than 2 per 10 high-power fields. Grade 2 (G2) are well-differentiated intermediate-grade pNETs having a Ki-67 index of 3–20% of ≥500 cells and/or a mitotic count of 2–20 per 10 high-power fields. Grade 3 (G3) includes high-grade tumors presenting a Ki-67 index over 20% of ≥500 cells and/or a mitotic count over 20 per 10 high-power fields. Inclusion in the G3 group is based only on the values of Ki-67 index and/or mitotic count, regardless of the other morphological features of tumors. G3 contains two subgroups of cancers which, despite their similar proliferation rate, are different for both their malignancy degree and their genetic background: (1) well-differentiated high-grade pNETs with round homogeneous nuclei, low-to-moderate atypia, salt and pepper-like chromatin, and fine granular cytoplasm, with frequent abnormalities of the MEN1 tumor suppressor gene (25–44%), the apoptotic regulator Death Domain-Associated Protein (DAXX) gene (25%), or the chromatin modifier Alpha-Thalassemia/mental Retardation X-linked (ATRX) gene (18%); (2) poorly-differentiated high-grade pNECs (small cell type and large cell type), which usually have an extremely high mitotic rate (over 50%) and are commonly mutated on the Retinoblastoma (RB) or the Tumoral Protein 53 (TP53) tumor suppressor genes. The different genetic bases demonstrate that G3-pNECs do not derive from a malignant progression of well-differentiated pNETs but develop as separate tumors with a more severe grade of malignancy.
Investigation of the molecular pathogenesis of pNETs is limited by their rarity, multiple oncological and endocrinological outcomes, and heterogeneous genetics.
2. pNETs in Multiple Endocrine Neoplasia Type 1
pNETs arise in 30–80% of patients with Multiple Endocrine Neoplasia type 1 (MEN1), a rare inherited cancer syndrome characterized by the development of multiple neuroendocrine and nonendocrine tumors in a single patient and caused by a germline heterozygote loss-of-function mutation of the MEN1 tumor suppressor gene.
A great majority of MEN1 patients develop, during their life, multiple pancreatic microadenomas (less than 0.5 cm in diameter), mostly NF-pNETs (approximately 50% of cases) [4], insulinomas (7–31% of cases) [5], and gastrinomas (about 5% of cases) [6]. Solitary pNETs in MEN1 manifest less frequently (less than 13% of cases), usually as large masses (macroadenomas > 2 cm), and are often nonfunctional [7]. MEN1 pNETs manifest at a younger mean age compared to their sporadic counterparts (10–50 years vs. 50–80 years). MEN1 gastrinomas are prevalently located within the duodenum (over 90% of gastrinomas), and less than 10% of cases affect the pancreas. Glucagonomas, somatostatinomas and vasoactive intestinal polypeptide secreting tumors (VIPomas) manifest less frequently (3–4%, <2% and <2%, respectively) [5]. Neuroendocrine tumors affecting the gastro-entero-pancreatic tract in MEN1 syndrome are shown in Figure 1.
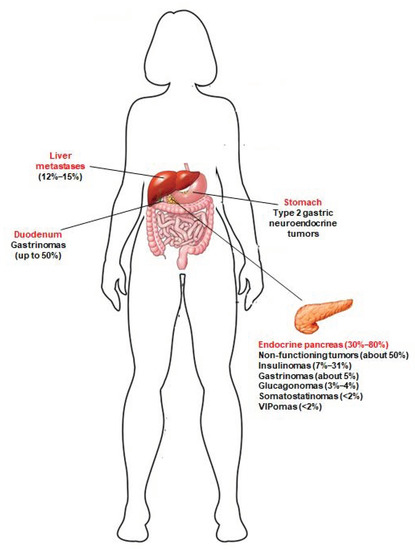
Figure 1. Neuroendocrine tumors of the gastro-entero-pancreatic tract associated with MEN1 syndrome. Prevalence of these tumors in MEN1 patients is indicated between brackets. Type 2 gastric neuroendocrine tumors develop in MEN1 patients, as a consequence of Zollinster–Ellison syndrome (ZES) from duodenal and/or pancreatic gastrinomas; to date, little is known about the true prevalence of these gastric neuroendocrine lesions in MEN1 patients. Jensen et al. [5] indicated a prevalence of 7–35%. Recently, Manoharan et al. [8] demonstrated that type 2 gastric neuroendocrine tumors occur only in MEN1 patients with ZES, and less frequently than previously reported (5.3% of the total of MEN1 patients and 12.5% of MEN1 patients with ZES).
Secreting pNETs are usually associated with distinct endocrine syndromes, caused by the excessive amount of hormone released by tumor cells. As their sporadic counterpart, MEN1 pNETs result positive to immunostaining for neuroendocrine markers and, in case of functioning tumors, their specific secreted hormones.
MEN1 pNETs follow the same three-grade WHO classification and they usually present a slow growth rate, with an estimated doubling time of 5–10 years [9]. However, in some cases, MEN1-associate pancreatic tumors exhibit a more aggressive behavior than the sporadic forms, with metastasis described in about 23–33% of cases. Tumor size is an important predictive factor of prognosis and overall survival in MEN1 pNETs, mostly for NF-pNETs; tumors larger than 2 cm in diameter have been associated with a high risk of malignancy and metastases development. Even if characterized by a slow disease progression, when compared with the non-endocrine pancreatic cancers and their endocrine sporadic counterpart, and despite the availability of an early genetic and clinical diagnosis and the possibility of praecox surgical intervention, pNETs currently remain the main cause of death among MEN1 patients, with an estimated 10-year survival after diagnosis of NF-pNETs ranging from 23% to 62% [10].
Currently, no prognostic factors allow clear identification of MEN1 patients at risk of metastatic cancers, nor to tailor personalized pNET treatment. KI-67 labeling and mitotic count have both been shown to be prognostic in sporadic pNETs [11][12], but few data are available for MEN1 pNETs. A study by Conemans et al. [13] evaluated, in formalin-fixed-paraffin-embedded pNET tissues from 69 MEN1 patients, the prognostic value of Ki-67 labeling index and mitotic rate, according to the WHO grade, with respect to clinical features of the tumors (i.e., tumor size, presence/absence of liver metastases, and time of metastases diagnosis after identification of the primary tumor). They showed that the mitotic count value was associated with the development of metastases only for NF-pNETs, but not for insulinomas, with 80% of metastases occurring in the G2 cancers (mitotic rate 2–20 per 10 high-power fields) and only in 23% of G1 tumors (mitotic rate <2 per 10 high-power fields). No significant association was found between values of the Ki-67 labeling index and metastases development. Moreover, the authors confirmed that tumor size is the main risk factor associated with liver metastases, both for insulinomas and NF-pNETs (in their cohort, the cut off diameter of primary tumor causing metastases was 3 cm, with respect to the 2 cm previously indicated by the guidelines) [13].
Unfortunately, it is not possible to predict cancer type development, tumor behavior, or risk of metastases based on the MEN1 gene mutation. MEN1 patients have a variable clinical expressivity; clinical manifestations, including pNETs, can be different between members of the same family bearing the same MEN1 mutation, and even between homozygous twins, suggesting that other genetic, epigenetic, and environmental factors can affect MEN1 tumorigenesis and drive individual tumor development and progression.
References
- Lawrence, B.; Gustafsson, B.I.; Chan, A.; Svejda, B.; Kidd, M.; Modlin, I.M. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol. Metab. Clin. N. Am. 2011, 40, 1–18.
- Inzani, F.; Petrone, G.; Rindi, G. The New World Health Organization Classification for Pancreatic Neuroendocrine Neoplasia. Endocrinol. Metab. Clin. N. Am. 2018, 47, 463–470.
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188.
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170.
- Jensen, R.T.; Norton, J.A. Treatment of Pancreatic Neuroendocrine Tumors in Multiple Endocrine Neoplasia-Type 1(MEN1): Some Clarity but Continued Controversy. Pancreas 2017, 46, 589–594.
- Tonelli, F. Zollinger-Ellison Syndrome in Men1 Patients: Medical or Surgical Treatment? Ann. Surg. 2006, 244, 61–70.
- Kamilaris, C.D.C.; Stratakis, C.A. Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front. Endocrinol. 2019, 10, 339.
- Manoharan, J.; Anlauf, M.; Albers, M.B.; Denzer, U.W.; Mintziras, I.; Wächter, S.; di Fazio, P.; Bollmann, C.; Bartsch, D.K. Gastric enterochromaffin-like cell changes in multiple endocrine neoplasia type 1. Clin. Endocrinol. 2021.
- Kann, P.H.; Balakina, E.; Ivan, D.; Bartsch, D.K.; Meyer, S.; Klose, K.-J.; Behr, T.H.; Langer, P. Natural course of small, asymptomatic neuroendocrine pancreatic tumours in multiple endocrine neoplasia type 1: An endoscopic ultrasound imaging study. Endocr. Relat. Cancer 2006, 13, 1195–1202.
- Pea, A.; Hruban, R.H.; Wood, L.D. Genetics of pancreatic neuroendocrine tumors: Implications for the clinic. Expert. Rev. Gastroenterol. Hepatol. 2015, 9, 1407–1419.
- Ekeblad, S.; Skogseid, B.; Dunder, K.; Oberg, K.; Eriksson, B. Prognostic factors and survival in 324 patients with pancreatic endocrine tumor treated at a single institution. Clin. Cancer Res. 2008, 14, 7798–7803.
- Scarpa, A.; Mantovani, W.; Capelli, P.; Beghelli, S.; Boninsegna, L.; Bettini, R.; Panzuto, F.; Pederzoli, P.; delle Fave, G.; Falconi, M. Pancreatic endocrine tumors: Improved TNM staging and histopathological grading permit a clinically efficient prognostic stratification of patients. Mod. Pathol. 2010, 23, 824–833.
- Conemans, E.B.; Brosens, L.A.A.; Raicu-Ionita, G.M.; Pieterman, C.R.C.; de Herder, W.W.; Dekkers, O.M.; Hermus, A.R.; van der Horst-Schrivers, A.N.; Bisschop, P.H.; Havekes, B.; et al. Prognostic value of WHO grade in pancreatic neuro-endocrine tumors in Multiple Endocrine Neoplasia type 1: Results from the DutchMEN1 Study Group. Pancreatology 2017, 17, 766–772.

