 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | MARIALUIGIA FANTACUZZI | + 4857 word(s) | 4857 | 2021-04-20 08:54:09 | | | |
| 2 | Camila Xu | Meta information modification | 4857 | 2021-04-23 12:11:03 | | |
Video Upload Options
CDKs, a family of serine/threonine kinases, regulate cell cycle progression into the four distinct phases G1, S (DNA synthesis), G2 and M, and are crucially involved in the regulation of cell division and proliferation.
1. Introduction
Breast cancer (BC) is the most recurrent cancer in women worldwide, impacting 2.1 million women each year according to World Health Organization [1]. BC is a heterogeneous disease due to genetic factors that are reflected in different phenotypes. BC can be divided into different subtypes: luminal, in which estrogen receptors (ER) and/or progesterone receptors (PR) are expressed, further divided into luminal A and luminal B subtypes depending on the expression of Ki67 (low levels in A and high in B); HER2+, in which the human epidermal growth factor receptor 2 (HER2) is overexpressed and ER and PR are lacking; triple negative BC (TN), in which the previous targets are not expressed. The estrogen-receptor positive (ER+) BC is the most common type, with the prevalence of about 60% of cases in pre-menopausal women and 75% in post-menopausal women [2][3][4][5].
The anti-hormonal treatment involves the suppression or reduction of the estrogen effects and can be carried out using drugs that limit the production of these hormones, such as aromatase inhibitors (AIs), or act on the ER receptor, such as selective ER modulators (SERM) or down-regulators (SERD) [6][7][8]. The adjuvant therapy consists of 5–10 years of ER-directed endocrine therapy that result in a reduction of mortality in ER+ BC of more than 40%. Resistance to endocrine therapy leading to early-stage ER+ BC is common and decisive in the setting of advanced disease [9][10].
The aromatization reaction in the final step of estrogen biosynthesis is unique, therefore this reaction becomes an excellent target for inhibiting the synthesis of estrogens without affecting the production of other steroids. In recent decades, several aromatase inhibitors have been developed to adequately suppress estrogen production and have been used in the treatment of estrogen-dependent BC [11][12][13][14][15][16].
Given the high percentage of resistance to aromatase inhibitor treatment, new therapeutic strategies have been identified to make the treatment of ER+ BC more effective. One of the mechanisms involved in the resistance concerns the activation of cyclin-dependent kinases (CDKs) as an ER-independent growth signal, that involves the important protein kinase signaling pathway (PI3K/AKT/mTOR) (Figure 1) [17][18].
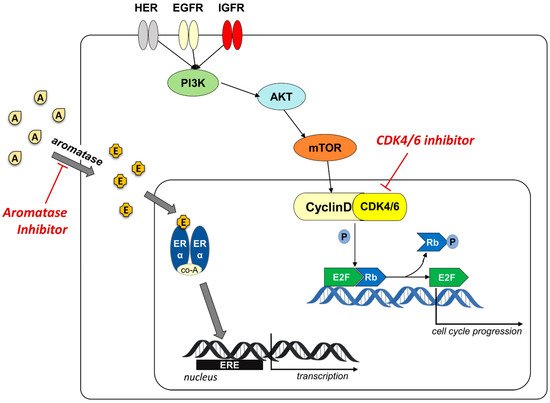
Figure 1. Schematic representation of the cyclinD/CDK4/6 involvement in overpassing resistance to aromatase inhibitors. Aromatase converts androgens (A) in estrogens (E) that bind to ER receptor. The recruitment of co-activators (co-A) allows the binding to ERE element on the target genes and the activation of the transcription. AIs block the production of estrogen inhibiting the ER-driven activation of cell cycle progression. The activation of cyclinD/CDK4/6 complex, mediated by the protein kinase signaling pathway (PI3K/AKT/mTOR), stimulates cell proliferation independently from aromatase. The use of CDK4/6 inhibitor blocks this alternative activation pathway.
CDKs, a family of serine/threonine kinases, regulate cell cycle progression into the four distinct phases G1, S (DNA synthesis), G2 and M, and are crucially involved in the regulation of cell division and proliferation. CDK stability, activation and downstream phosphorylation is controlled by cyclin counterpart and endogenous inhibitors. To date, 21 CDKs are known and their role in different types of cancer has been reported by many research groups [19][20][21]. In particular, CDK1, 2 and 4 regulate the transition of the cell cycle steps, CDK 7, 8, 9 and 11 regulate the gene transcription, while CDK 6 regulates both [22][23][24].
Mitogenic, hormonal, and growth factors allow the cyclin D to bind CDK4/6, forming the complex that regulates the phosphorylation status of retinoblastoma protein (Rb). The phosphorylated-Rb determines the dissociation of the transcription factor E2F that binds to DNA and promotes the expression of different genes, regulates DNA replication and cell division, with the transition from G1 to S phase (Figure 1). Several endogenous factors control cell proliferation, including INK4 family proteins (p16, p15, p18, p19), cyclin inhibitory proteins and kinase inhibitory proteins (KIPs, p21, p27) [25][26].
The dysregulation of cell cycle caused by the overexpression or gain-of-function mutations in the CDKs and cyclins or the loss of endogenous inhibitors expression and function, is recurrent in many cancer diseases. In BC, but also in other type of cancer, a dysregulation of this process leads to a proliferative stimulus, and a significant role in this mechanism is played by the overexpression of cyclin D. Inhibition of cell cycle using CDK4/6 inhibitor has emerged as antitumor treatment in BC in association to the hormonal therapy to overpass resistance to AIs and avoiding relapses [27][28][29][30][31][32].
Over the years several CDK inhibitors have been developed and tested in different types of cancer [33][34][35][36][37]. First generation inhibitors (flavopiridol and roscovitine) demonstrated an inadequate balance between efficacy and toxicity, due to their action on several kinases (pan-inhibitors) [38][39][40]. Second generation inhibitors (dinaciclib) were developed with the aim to increase selectivity and potency, but demonstrated limited efficacy and considerable toxicity in clinical studies. The toxicity of these compounds is associated with the multi-target activity against isoforms fundamental for the proliferation (CDK1) and survival (CDK9) of normal cells [41][42][43][44]. However, in recent years, the interest in identifying inhibitors of specific kinases with targeted action on tumor cells and with less toxic effects, has led to the discovery of selective CDK4/6 inhibitors [30][45]. Third generation CDK inhibitors selectively inhibit CDK4/6 with potent efficacy and reduced toxicity, such as the FDA approved palbociclib (1), ribociclib (2), and abemaciclib (3) (Figure 2) [46][47][48][49].
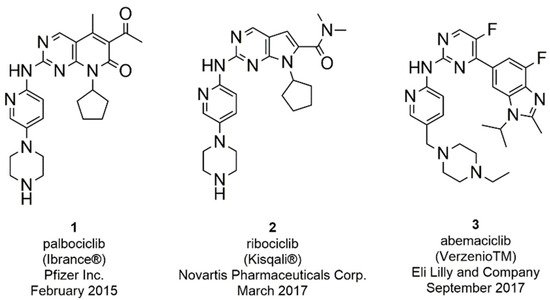
Figure 2. FDA approved CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib.
The search for even more selective CDK4/6 inhibitors is still a challenge in the development of novel anticancer therapies. To date, three ATP-competitive CDK 4/6 selective inhibitors (trilaciclib, lerociclib and SHR-6390) are under advanced clinical trials. Trilaciclib (G1T28, 4, Figure 3) is currently ongoing phase II trials in patients with small cell lung cancer (NCT02514447, NTC02499770) and with hormone receptor negative BC (NCT02978716), while entered in phase III for metastatic colorectal cancer (NCT04607668) [50][51][52][53]. Phase II (NCT02983071) trial of lerocliclib (G1T38, 5, Figure 3) examined the effect in combination with fulvestrant in hormone receptor-positive, HER2-negative locally advanced metastatic BC while another phase II study combined lerociclib with osimertinib in EGFR-mutant non-small cell lung cancer (NCT03455829) [54][55]. SHR-6390 (6, Figure 3) is currently ongoing phase II trial in patients with hormone receptor positive, ErbB2 negative BC (NCT03966898) [56][57]. Moreover, an abemaciclib related compound, BPI-16350 (7, Figure 3), recently entered phase I clinical trial (NCT03791112) in patients with advanced solid tumor and the estimated study completion date is in December 2021 [58].
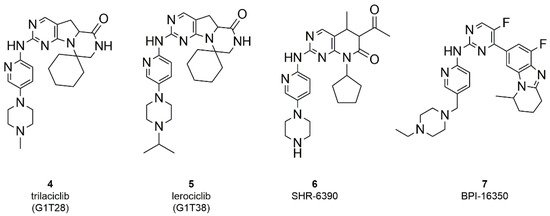
Figure 3. Chemical structure of ATP-competitive CDK 4/6 selective inhibitors in clinical trials.
Based on their mode of action, kinase inhibitors can be divided into two types: ATP-competitive and non-competitive inhibitors. The reported CDK4/6 small molecule inhibitors abemaciclib, palbociclib and ribociclib are ATP-competitive inhibitors, forming hydrogen bonds in the ATP binding site with the kinase ‘‘hinge” residues (Val101 in CDK6) and hydrophobic interactions in the region normally occupied by the ATP adenine ring (Figure 4A,C,D) [59][60][61].
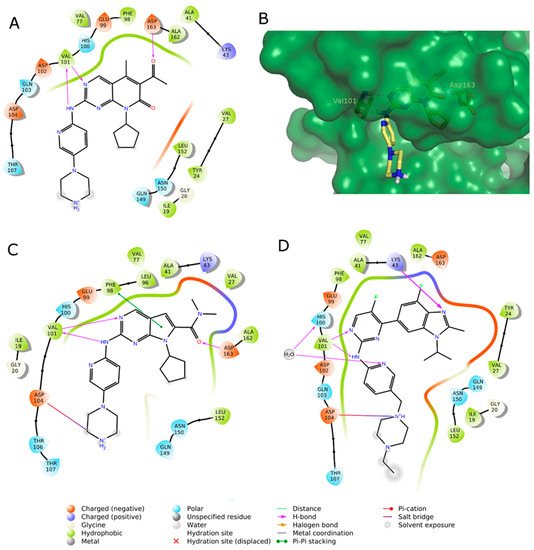
Figure 4. (A,C,D) 2D interaction diagram of the three approved CDK4/6 selective inhibitors as observed in their X-ray complexes into CDK6: (A) palbociclib (PDB ID: 5L2I); (C) ribociclib (PDB ID: 5L2T); (D) abemaciclib (PDB ID: 5L2S). (B) 3D representation of the X-ray binding geometry of palbociclib (stick, yellow C atoms) into the CDK6 binding site (dark green solid surface). Protein residues involved in key H-bond interactions are represented as stick; H-bonds are depicted as magenta dashed lines.
According to the study of Chen et al., the interactions enhancing the CDK inhibition and selectivity over other kinases are the H-bonds with the sidechains of His100 and Lys43, as shown for abemaciclib (Figure 4B) and a solvent-mediated interaction of a positively charged atom of the ligand and a solvent-exposed ridge consisting of Asp104 and Thr107 [62].
2. Small-Molecule Inhibitors
Small molecule inhibitors (SMIs) are compounds of ≤500 Da size, often administered orally, useful in cancer diseases. The traditional chemotherapy includes single or combination-therapy of drug targeting dividing tumor cells. The principal drawback of this non-targeted approach is the non-selective action on both normal and cancer cells. SMIs bind to specific molecular targets (targeted therapy) and selectively eliminate malignant cells [63][64]. The small size allows to use these molecules towards extracellular, surface, or intracellular proteins, including anti-apoptotic proteins that play a key role in cell growth and promotion of metastases. The most interesting targets in the antitumor field are mainly kinases such as serine/threonine/tyrosine kinases, matrix metalloproteinases (MMP), and CDKs [63][64][65][66][67][68].
The following SMIs are classified based on their chemical scaffold.
2.1. Thiazolyl-Pyrimidine Derivatives
Starting from the structure of abemaciclib and considering the fundamental interactions with the hinge region of CDK4/6, Tadesse et al. synthesized three series of compounds keeping constant pyrimidine, pyridine, and the amino linker to maintain the coplanarity of the two rings for ATP-mimetic kinase inhibitors (Figure 5), that represents the characteristic moiety of the three approved inhibitors [69].
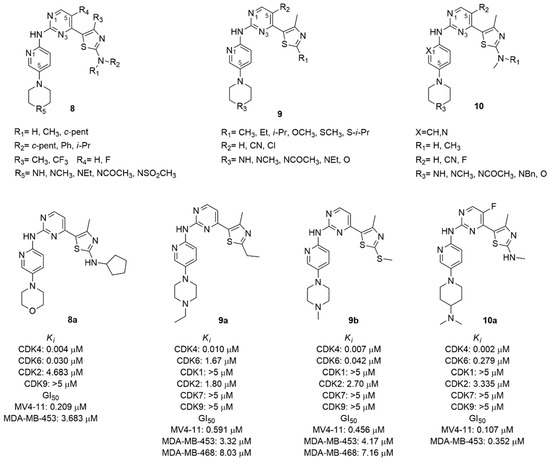
Figure 5. CDK4/6 inhibitors based on thiazolyl-pyrimidine scaffold.
The thiazole C2 amino moiety, introduced in previously synthesized CDK9 inhibitors, established a H-bond with the highly conserved Asp (Asp163 in the CDK6) residue among CDKs family of the Asp-Phe-Gly motif in the ATP-binding pocket, improving strong hydrophobic interaction of the methyl thiazole with the gatekeeper residues [70]. For this reason, the 2-amine-thiazole was introduced in C4 of the pyrimidine scaffold. The pharmacophore of the Tadesse’s lead compound is the N-(5-(piperidin-1-yl)pyridin-2-yl)-4-(thiazol-5-yl)pyrimidine (8–10, Figure 5). The main modifications of the first series (8, Figure 5) concerned the mono or di-substitution of the amine group (R1, R2) with methyl, ciclopentyl, phenyl or iso-propyl; the introduction of a fluorine atom in C5 of pyrimidine (R4) to optimize the pharmacokinetic properties; the N4 (R5) of the piperazine was substituted with a variety of ionizable groups. The most active compound (8a, Figure 5) contains a morpholine ring and a cyclopentyl substitution of the amine (Ki CDK4 = 0.004 μM, Ki CDK6 = 0.030 μM). Its antiproliferative activity was assessed by MTT assay in human leukemia Rb positive MV4-11 (GI50 = 0.209 μM), and in human breast cancer Rb negative MDA-MB-453 (GI50 = 3.683 μM) cell lines. The SAR analysis shows that the amino group in the thiazole ring could accept only a mono-substitution and the cyclopentyl is better than alkyl chain or aromatic ring; the introduction of the electron withdrawing trifluoromethyl increases the toxicity; the substitution of the second nitrogen atom of the piperazine with carbon and the exocyclic primary amino decreases activity and selectivity, while the introduction of oxygen increases the selectivity toward CDK4/6.
In the second series of derivatives (9, Figure 5), the amino group on thiazole ring was replaced with alkyl, ether or thioether substituents, a cyano group or chlorine atom on C5 of pyrimidine was introduced, and the C4 of piperidine was replaced by an oxygen atom or secondary or tertiary amine substituted with alkyl or acetyl group [71]. Among the synthesized compounds, 9a and 9b (Figure 5) emerged with good CDK inhibition values (Ki CDK4 = 0.010 and 0.007 μM, Ki CDK6 = 1.67 and 0.042 μM, respectively) and antiproliferative activity in MV4–11 (GI50 = 0.591 μM and 0.456 μM, respectively). The antiproliferative activity of compounds 9a–b was also evaluated in a panel of human cancer cell lines including breast, colon, ovary, pancreas, prostate, leukemia and melanoma. Noteworthy, the different antiproliferative activity in the MDA-MB-453 (GI50 = 3.32 μM and 4.17 μM, respectively) and the corrensponding Rb-deficient MDA-468 (GI50 = 8.03 μM and 7.16 μM, respectively) confirms the mechanism of action of these compounds which act in the presence of the intact Rb function. The potent effect on the growth of melanoma M249 (GI50 = 0.47 μM and 0.91 μM, respectively) and of resistant to dabrafenib M249R (GI50 = 0.27 μM and 0.91 μM, respectively) cell lines paves the way for a possible therapeutic use in melanoma. The SAR analysis highlighted that the presence of the nitrogen atom of the pyridine is fundamental for CDK4/6 selectivity, and the substitution of the amine group in C2 of the thiazole with alkyl, thioether or ether prevents the H-bond with the conserved Asp163, increasing the selectivity.
In another series of derivatives (10, Figure 5) the substitution of the pyridine with a benzene ring, the mono-substitution of the amino group of the thiazole, the introduction of fluorine atom or cyano group in C5 of pyrimidine, and the alkylation of the piperidine nitrogen or the introduction of morpholine or piperidine were studied [72]. Compound 10a (Figure 5) was the most active in biological assays, with good CDK 4/6 inhibition (Ki CDK4 = 0.002 μM, Ki CDK6 = 0.279 μM), selectivity over other kinases and good pharmacokinetic profile. The antiproliferative activity was tested in different cancer cell lines such as leukemia or solid cancers (breast, colorectal, melanoma, ovarian, prostate).
2.2. Benzimidazolyl-Pyrimidine Derivatives
Zha et al. focused their attention to the substitution of the abemaciclib pyridine with a benzoimidazolyl in C4 and the introduction of tetrahydro-naphthyridine as substituent of the amino linker, with the aim to discover conformationally restricted analogs sharing improved activity and selectivity [73]. Compound 11 (Figure 6), discovered through the combination of structure-based drug design and traditional medicinal chemistry approaches, retains all key contacts between abemaciclib and CDK6, such as the edge-to-face interaction of the benzimidazole ring and Phe98 of the gatekeeper and the H-bonds of the amino pyrimidine with residues in the hinge loop. The tetrahydro-naphthyridine forms an additional water-mediated H-bond between the aromatic nitrogen with His100 and a salt-bridge interaction of the physiologically protonated nitrogen and Asp104 (Figure 4D) [19][74].
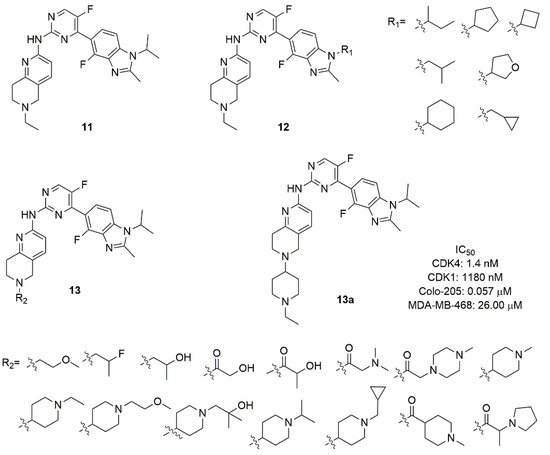
Figure 6. CDK4/6 inhibitors based on benzimidazolyl-pyrimidine scaffold.
The authors explored the removal of the nitrogen in pyridine, in 2,4-pyrimidine or the substitution with a 4,6-pyrimidine, confirming that the nitrogen atoms are fundamental for CDK6 selectivity over CDK1. Although compound 11 exhibited good activity (IC50 CDK4 = 1.5 nM) and a selectivity index of 311, the very poor pharmacokinetic properties required further optimization. In compounds of the series 12 (Figure 6) a cyclic or acyclic alkyl substitution of the iso-propyl of the imidazole of compound 11 was introduced without a substantial improvement of activity, suggesting that the hydrophobic cleft of the protein could not host rigid or bulky groups. Compounds of the series 13 (Figure 6) contain a variety of substituents on the protonable nitrogen of tetrahydro-naphthyridine. The hydrophilic substitutions maintain the inhibition potency, with amide analogues suffering from poor exposure; the introduction of a N-alkyl piperidine improves the inhibitory activity and selectivity. In fact, compound 13a (Figure 6) emerged as the best compound, with enzymatic CDK4 IC50 of 1.4 nM and selectivity CDK1/CDK4 around 850 (IC50 CDK1 = 1180 nM), good antiproliferative activity in Colo-205 cell line (IC50 = 0.057 μM), favourable in vitro metabolic properties (microsomal stability and CYP isoforms inhibition) and robust pharmacokinetic properties in mice and rats.
Wang et al. synthesized and tested a library of abemaciclib analogs [75]. The tetracycle scaffold (piperazine, pyridine, pyrimidine and benzimidazole) was kept constant, while small substituents were introduced on piperazine nitrogen (ethyl, 2-fluorethyl, cyclopropyl, iso-propyl), in C6 of pyridine (methyl), in C5 of pyrimidine (fluorine), and in C4 of benzimidazole (fluorine). The benzimidazole ring was transformed in a tricycle by connecting N1 and C2, inserting a cyclopentyl, cyclohexyl or cycloheptene [75].
A large library of 23 compounds (14, Figure 7) was tested for CDK1 and CDK4/6 activity. Compounds demonstrated null activity versus CDK1, but a remarkable inhibition of CDK4/6, with IC50 ranging from 0.6 to 340 nM. Compound 14a (IC50 CDK4 = 7.4 nM and IC50 CDK6 = 0.9 nM) was further tested for hERG channel inhibition, showing low heart toxicity. The pharmacokinetic parameters of 14a (Cmax, AUC, T1/2, MRT, CL/f, V2/F) demonstrated drug-like properties for following development. A docking study revealed that the di-methyl cyclopentyl group contributed to favourable H-bond between the nitrogen atom of the imidazole-condensed cycle and the amine group of Lys43.
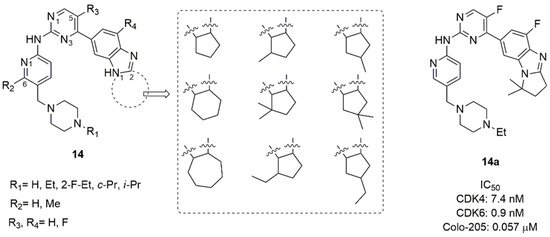
Figure 7. CDK4/6 inhibitors based on benzimidazolyl-cycloalkyl-pyrimidine scaffold.
The studies on Colo-205 subcutaneous xenografts tumor model in BALB/c nude mice revealed that compound 14a was not well tolerated and had a narrow therapeutic window [73]. For this reason, Shi’s research group synthesized two novel series of benzimidazolyl-pyrimidine containing the tetrahydro-naphthyridines with a dimethylamino-ethyl group as substituent on protonable nitrogen of the bicycle: in the series 15 (Figure 8) the nitrogen atom of imidazole was substituted with alkyl or cycloalkyl, while in the series 16 (Figure 8) the protonable nitrogen of the ethylamine chain was substituted [76]. Compound 16a (Figure 8) demonstrated nanomolar in vitro activity (IC50 CDK4 = 0.71 nM, IC50 CDK6 = 1.10 nM) with high kinase selectivity, excellent metabolic properties, good pharmacokinetic properties, low toxicity, and desirable antitumor efficacy in MCF-7, Colo-205, and A549 xenograft murine models. Even though compounds of series 16 possessed fair CDK4/6 activity in the range of nanomolar (IC50 = 0.999–6.14 nM), many of them failed in antiproliferative activity in MCF-7, T-47D, ZR-75-1, and Colo-205 cell lines.
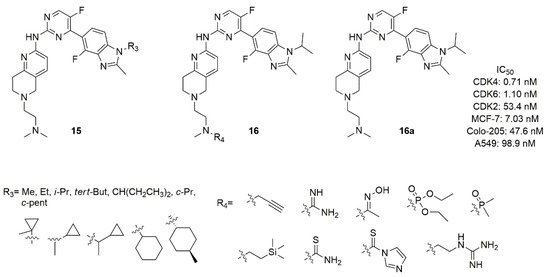
Figure 8. Structure of Shi’s research group of benzimidazolyl-pyrimidine derivatives.
The SAR analysis demonstrated that, with respect to N-iso-propyl (16a), the N-methyl or N-ethyl substitution of the imidazole decreased the activity more than others alkyl or cycloalkyl groups in terms of CDK 4/6 activity. The cycloalkyl, probably for its steric hindrance, also decreases the antiproliferative activities. Considering the substitution of N-methyl on the nitrogen of the ethylamine chain on tetrahydro-naphthyridine, the introduction of a bulkier group was unproductive in terms of CDK4/6 activity and selectivity over CDK2.
2.3. Pyrido-Pyrimidine Derivatives
Considering the pyrido [2,3-d]pyrimidine scaffold of palbociclib, Abbas et al. synthesized two series of 7-thienylpyrido[2,3-d]pyrimidines (17 and 18, Figure 9) and tested them for CDK6 inhibition and cytotoxicity against breast, lung, and prostate cancer cell lines [77]. In the series 17, the 2-aryldiene hydrazinyl moiety was introduced in the scaffold and the aryl group was substituted in para-position. The most active compound of this series resulted 17a, containing a para-methoxy group on the benzene ring. In fact, it demonstrated a CDK6 IC50 value of 115.38 nM and a good cytotoxicity against breast MCF-7 (IC50 = 1.59 μM), prostate PC-3 (IC50 = 0.01 μM), lung A-549 (IC50 = 2.48 μM) cancer cells.
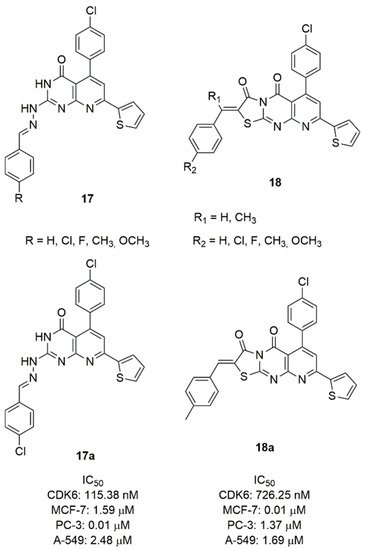
Figure 9. CDK6 inhibitors based on the pyrido-pyrimidine scaffold.
The series 18 is constituted by a fused ring to pyrimidine forming a tricyclic pyridothyazolopyrimidine, substituted in C2 with a para-substituted benzylidene. Compound 18a emerged as the most potent CDK6 inhibitor (IC50 = 726.25 nM) and cytotoxic against MCF-7 (IC50 = 0.01 μM), prostate PC-3 (IC50 = 1.37 μM), lung A-549 (IC50 = 1.69 μM) cancer cell lines.
2.4. Imidazo-Pyrido-Pyrimidine Derivatives
The pyrido-pyrimidine scaffold of palbociclib was fused with imidazole in two novel series of CDK4/6 inhibitors containing the fused tricyclic ring of imidazo[10,2′:1,6]pyrido[2,3-d]pyrimidine (19 and 20, Figure 10) [78].
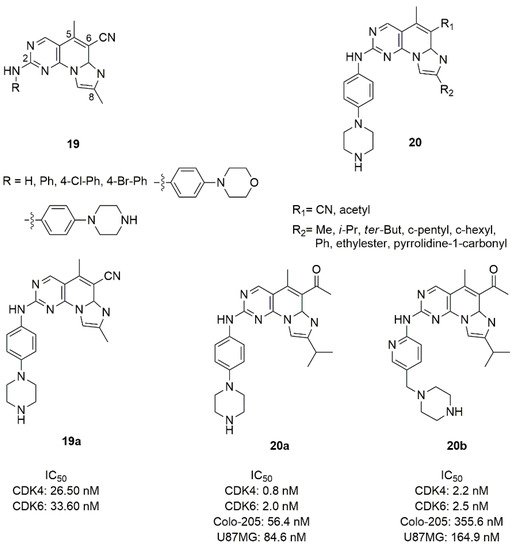
Figure 10. Structure of imidazo[10,2′:1,6]pyrido[2,3-d]pyrimidine derivatives.
In the series 19, the fused tri-heteroaryl structure was substituted with a methyl in C5 and C8, cyano in C6, while the amino group in C2 was substituted by phenyl or para-substituted phenyl groups. This type of modification did not sufficiently improve the activity, and compound 19a with the piperazine in para position showed modest inhibition values (IC50 CDK4 = 26.50 nM, IC50 CDK6 = 33.60 nM). Keeping constant the piperazine moiety, in the series 20 the C6 and C8 positions were changed by the introduction of methyl, iso-propyl, terz-butyl, cyclopentyl, cyclohexyl, phenyl, ethylester, or pyrrolidine-1-carbonyl in C8, while the cyano group in C6 was replaced with the acetyl one. Compound 20a was the best one of the series in terms of inhibition (CDK4 IC50 = 0.8 nM, CDK6 IC50 = 2.0 nM).
The piperazine ring of compound 20a was also replaced by saturated heterocycle, distanced by a methyl and a carbonyl linker; alternatively the piperazinyl-pyridine portion was replaced with a fused bicycle. None of these changes improved the inhibition of kinases [79]. Compound 20a demonstrated good activities on Colo-205 (IC50 = 56.4 nM), and glioma U87MG (IC50 = 84.6 nM) cell lines, favourable in vitro metabolic properties (microsomal stability, CYP isoforms inhibition), acceptable pharmacokinetic profiles in mice and rats, antitumor efficacy with controllable observed side effects in xenograft in vivo studies.
2.5. Pyrazolo-Quinazoline Derivatives
Considering the inhibition activity of different kinases (Aurora-A, CDK2, Polo-like Kinase 1) of the 4,4-dimethyl-4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline [80], Zhao et al. synthesized a series of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazolines (21, Figure 11) and tested their inhibition of CDK4/6 [79]. The amine group in C2 position of the quinazoline was substituted with pyridine or benzene ring. Compounds containing the pyridine confirmed that the nitrogen atom in this position affects not only the inhibitory activity, but also the cellular activity against MCF-7 cell line. In fact, the pyridine derivatives were more active as CDK4/6 inhibitors and displayed improved cellular activity.
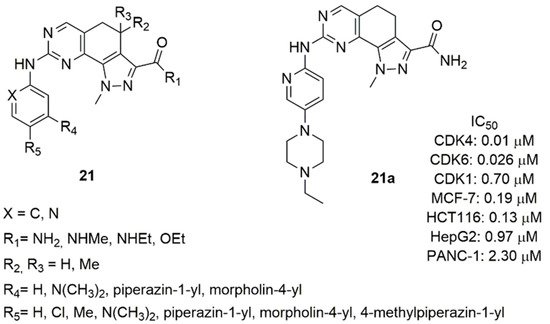
Figure 11. Structure of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives.
Compound 21a was the best one of this series, showing good activity on CDK4/6 (IC50 CDK4 = 0.01 μM, IC50 CDK6 = 0.026 μM) and high selectivity against CDK2 (IC50 CDK2 = 0.70 μM), anti-proliferative activity in MCF-7 cell line (IC50 = 0.19 μM) and other solid tumors (colorectal, liver, pancreatic), favorable pharmacokinetic parameters (T1/2, CL, AUC, V, Cmax).
3. PROTACS
The therapeutic use of small-molecule inhibitors to target proteins such as transcription factors, non-enzymatic, and scaffolding proteins, has several limitations because these targets lack appropriate active site to be occupied that directly modulate protein functions [81]. Moreover, high systemic drug exposures in the use of small molecules that bind to the active site of a protein are required to achieve site occupation, which may lead to an increase in adverse effects caused by binding to off-target sites [82]. Other complications in the prolonged use of small molecule inhibitors are the possible mutation of the target protein and the establishment of resistance to the therapy, the overexpression of such protein to balance the inhibition drug-mediated, and the accumulation. These mechanisms are associated with the partial or overall suppression of the downstream signaling pathways [83].
A strategy to circumvent the problem of binding site occupancy to regulate the inhibition of a protein and the possibility of significantly expand the number of proteins that can be inhibited, is the use of small-molecule-induced protein degradation. In this way, the pharmaceutical advantages deriving from the use of small molecules are preserved and the proteins generally considered “undraggable” are removed [84]. These hybrid molecules, generally called PROteolysis-TArgeting Chimeras (PROTACs), are constituted by two small binding molecules connected by a linker (Figure 12): one domain is directed to the targeted protein, while the other domain binds E3 ubiquitin ligase. The complex allows the binding of the proteolytic ubiquitin on the target protein, and its consequent degradation of the targeted protein in proteasome. PROTACs act catalytically and are not destroyed as small molecule suicide inhibitors that permanently bind target macromolecules [85][86].
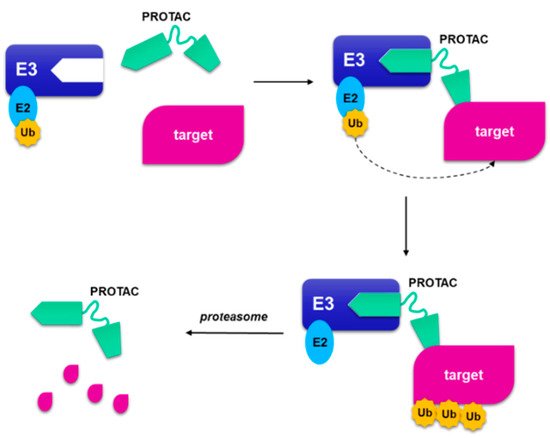
Figure 12. A schematic representation of proteolysis targeting chimera. The PROteolysis-TArgeting Chimera (PROTAC) is composed by a portion that binds to the ubiquitin ligase and a small molecule that binds to target protein, joined by a linker. When the targeted protein binds the small molecule, and the other part binds to E3 ligase, a ternary complex is formed. The following poliubiquitination of the target allows the proteasome to degrade the target protein and regenerate the PROTAC.
PROTAC strategy is widely applied to degrade proteins related to immune disorders, neurodegenerative diseases, viral infections, and cancer diseases [87][88][89]. In this paragraph the application of PROTAC strategy to CDK4/6 inhibitors is summarized.
The use of this approach could be exploited to selectively inhibit CDK6 with respect to CDK4, which have specific functions, could derive from the use of PROTACs. In fact, the binding site of ATP in kinases 4 and 6 possesses a high structural similarity, that could hardly be circumvented with the use of small molecules. All the reviewed studies have in common the binding of the E3-binding portion (E3 ligase ligand) to the nitrogen atom of the piperazine of the three approved CDK4/6 inhibitors. In fact, the crystallographic studies of palbociclib, ribociclib and abemaciclib show that the piperazine ring is projected towards the solvent (Figure 4B), in an optimal position to act as an anchor point.
Among the first studies reporting a PROTAC active towards CDK4/6, emerges the work of Zhao and Burgess [90], who combined palbocilib and ribocilib with pomalidomide (cereblon (CRBN), E3 ligase ligand) by means of a linker containing a triazole ring (22a–b, Figure 13). Studies on MDA-MB-231, a triple negative breast cancer cell line, showed that CDK4 is degraded more efficiently and PROTAC containing palbociclib (22a) is more potent (DC50 CDK4 = 12.9 nM, DC50 CDK6 = 34.1 nM) than 22b (DC50 CDK4 = 97 nM, DC50 CDK6 = 300 nM). The same CDK degradation and cytotoxicity studies conducted on MCF-7 showed that 22a–b are less efficient towards this cell line with respect to the triple negative cell line.
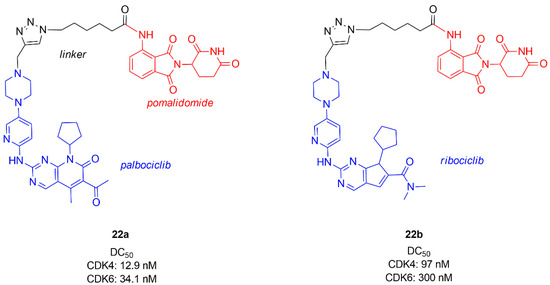
Figure 13. Chemical structures of palbociclib or ribociclib/pomalidomide PROTACs.
In the same period, Rana et al. synthesized a chimera series of palbociclib and pomalidomide by changing the length and the composition of the flexible linker (23, Figure 14) [91].
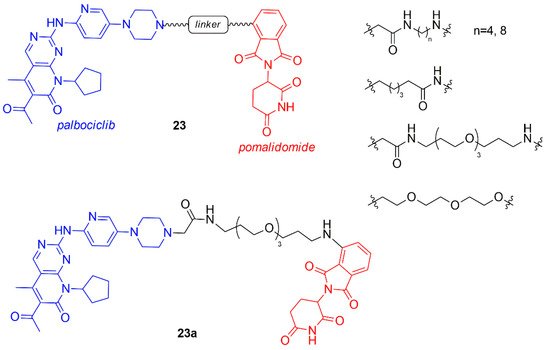
Figure 14. Chemical structures of palbociclib/pomalidomide PROTACs.
All compounds with shorter linker degrade CDK6 partly, while the PROTAC containing the longest linker (23a, Figure 14) selectively degraded CDK6 at the single dose of 500 nM in pancreatic cancer MiaPaCa2 cells with respect to other cyclin-dependent kinases including CDK4. Two hypotheses on the selective behavior of this PROTAC could be found in the less stable ternary complex palbociclib-E3 ligase-CDK4, that avoids the degradation, or in the fast deubiquitination of CDK4. The quantitative degradation of only CDK6 (CDK4 was not affected) was observed for compound 23a in a dose-response study at 4 and 24 h in Human Pancreatic Nestin-Expressing ductal (HPNE) and MiaPaCa2 cells at 100 nM.
Another library of PROTACs containing CDK4/6 inhibitor and pomalidomide was synthesized by Su et al. (24, Figure 15), in which the influence of the length and rigidity of the linker, the spatial orientation of the target protein and the E3 ligase, and the binding affinity of PROTAC to CDK4 and 6 were studied [92].
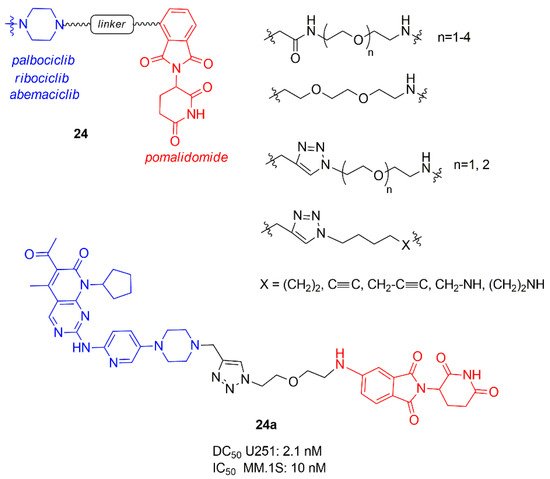
Figure 15. PROTACs containing CDK4/6 inhibitors and pomalidomide synthesized by Su et al.
PROTACs containing ribociclib did not degrade CDK6, while for the others best results in selectively degradation CDK6 was obtained with shorter linkers. In particular, the linker anchoring group to CDK inhibitors (amide, triazole, or methylene) did not influence the activity while to the other side, the best anchoring group to E3 ligase was the amino group, demonstrating that the flexibility of this portion is fundamental to correctly interact. The most potent PROTAC 24a possesses a DC50 value of 2.1 nM in glioblastoma U251 cells and demonstrated good potency also in hematopoietic cancer cells, including multiple myeloma MM.1S (IC50 10 nM).
Jiang and co-workers prepared a library of palbociclib, ribociclib, and abemaciclib PROTACs (25–27, Figure 16) connected to pomalidomide through an alkyl or polyethylene glycol (PEG) linker [93].
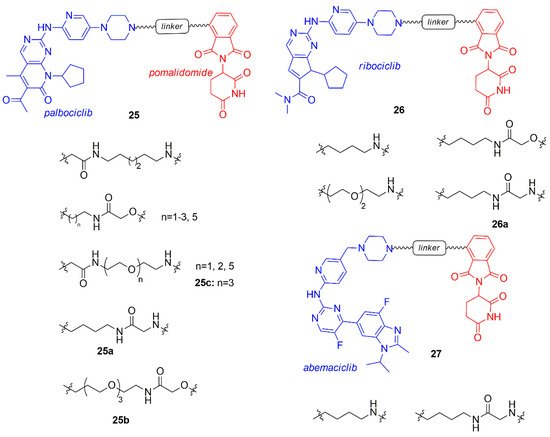
Figure 16. PROTACs containing CDK4/6 inhibitors and pomalidomide synthesized by Jiang et al.
PROTACs of each CDK4/6 inhibitor demonstrated degrading activity of both CDK4 and 6, but abemaciclib-PROTACs also induced the degradation of the off-target CDK9, that should be avoided [60]. The type of the linker (length and structure) and the CDK4/6 inhibitor of the PROTAC influenced the selectivity of degradation at 100 nM: compound 25a (alkyl linker conjugated to palbociclib) indifferently degraded both CDK4 and CDK6, 25b (extended PEG-3 linker conjugated to palbociclib) selectively hit CDK6, while 26a (4-carbon alkyl linker conjugated to ribociclib) was selectively toward CDK4.
Compounds containing the imide group were tested for their capability to inhibit Ikaros (IKZF1) and Aiolos (IKZF3), well-established targets of imide-based degraders [94][95][96]. Compounds 25a–b and 26a degraded also IKZF1/3, resulting in an enhnanced anti-proliferative effect on mantle cell lymphoma lines.
Compound 25c was previously synthesized by Brand and co-workers and studied for its ability to selectively degrade CDK6 over CDK4, in particular the correlation between the use of the CDK6 degrader in acute myeloid leukemia cells was investigated [97].
In a recent study, Anderson et al. evaluated the effect of other E3 ligase, such as von Hippel-Lindau (VHL) and Inhibitor of Apoptosis (IAP) instead of CRBN, on the selective degradation of CDK4/6, maintaining the anchoring on the nitrogen of piperazine of palbociclib and using different types of linkers (28, Figure 17) [98].
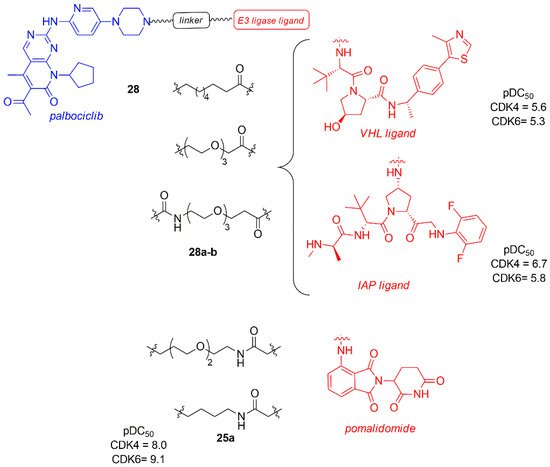
Figure 17. PROTACs palbociclib and E3 ligase ligands, such as von Hippel-Lindau (VHL) and Inhibitor of Apoptosis (IAP) ligands and pomalidomide.
The dose-response study in Jurkat cells after 24 h revealed that the degradation of CDK4 and CDK6 occurred independently of the type of E3 ligases (VHL, CRBN, and IAP binder), with a CDK4 pDC50 in the range of 6.2–8.0 and CDK6 pDC50 in the range of 7.7–9.1. It is important to note that all of them show a greater degradation power towards CDK6, probably due to a better stability of the formed ternary complex.
Compounds 28a–b, containing VHL and IAP, are the less potent degraders (28a: pDC50 CDK4 = 5.6; pDC50 CDK6 = 5.3; 28b: pDC50 CDK4 = 6.7; pDC50 CDK6 = 5.8), probably due to the linker nature. The most potent CDK4/6 degrader is 25a, previously reported by Jiang (pDC50 CDK4 = 8.0, pDC50 CDK6 = 9.1) [93].
Compounds 25a was taken into account by Steinbach et al. to synthesize novel palbociclib based PROTACs by changing the E3 ligase portion and inserting various linkers (29–31, Figure 18) [99]. In the series 29, pomalidomide was linked by an amide linker to the palbociclib piperidine, avoiding the protonation of the previously synthesized tertiary amine that could affect activity and selectivity. The linkers were polyethylene or alkyl chain of different size. These compounds were tested in multiple myeloma (MM.1S) cell lines at 0.1 μM and the activity of PROTACs was shown as the percentage of remaining CDK levels (D). The degradation percentage (D) of CDK6 for compounds 29a–c was in the range of 7.7–8.4 and the selectivity over CDK4 in the range of 1.9–3.3. In the series 30 and 31, palbociclib was linked to VHL ligand functionalized in two different positions to create an amide or a phenoxy group in the E3 ligase ligand side, while in the other side of the linker there was an amine group. Compound 30a showed a degradation percentage 1.7 and a selectivity CDK4 ratio of 19, while compound 31a showed a comparable degradation activity (D CDK6 = 1.4) but an improved selectivity (DCDK4/DCDK6 = 31). PROTACs 30a and 31a were also tested in different cancer cell lines (multiple mieloma, acute myeloid leukemia, acute lymphoid blastic leukemia) inhibiting cell proliferation.
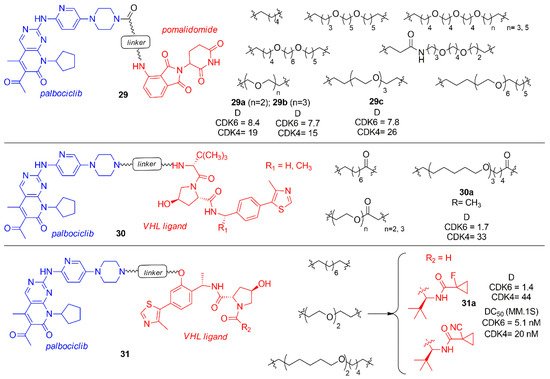
Figure 18. PROTACs containing palbociclib and E3 ligase ligand, such as pomalidomide and VHL ligand.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer. Res. 2015, 5, 2929–2943.
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genom. 2006, 7, 96.
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767.
- Tao, M.; Song, T.; Du, W.; Han, S.; Zuo, C.; Li, Y.; Wang, Y.; Yang, Z. Classifying breast cancer subtypes using multiple kernel learning based on bmics data. Genes 2019, 10, 200.
- Aggelis, V.; Johnston, S.R.D. Advances in endocrine-based therapies for estrogen receptor-positive metastatic breast cancer. Drugs 2019, 79, 1849–1866.
- Jordan, V.C.; O’Malley, B.W. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J. Clin. Oncol. 2007, 25, 5815–5824.
- Gombos, A. Selective oestrogen receptor degraders in breast cancer: A review and perspectives. Curr. Opin. Oncol. 2019, 31, 424–429.
- Early Breast Cancer Trialists’ Collaborative Group. Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352.
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer. 2019, 26, R15–R30.
- Dutta, U.; Pant, K. Aromatase inhibitors: Past, present and future in breast cancer therapy. Med. Oncol. 2008, 25, 113–124.
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase inhibitors: Role in postmenopausal breast cancer. Arch. Pharm. 2020, 353, e2000081.
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; et al. Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 2012, 55, 8464–8476.
- Fantacuzzi, M.; De Filippis, B.; Gallorini, M.; Ammazzalorso, A.; Giampietro, L.; Maccallini, C.; Aturki, Z.; Donati, E.; Ibrahim, R.S.; Shawky, E.; et al. Synthesis, biological evaluation, and docking study of indole arylsulfonamides as aromatase inhibitors. Eur. J. Med. Chem. 2020, 185, 111815.
- Di Matteo, M.; Ammazzalorso, A.; Andreoli, F.; Caffa, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Nencioni, A.; Parenti, M.D.; et al. Synthesis and biological characterization of 3-(imidazole-1-ylmethyl) piperidine sulfonamides as aromatase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3192–3194.
- Ammazzalorso, A.; Gallorini, M.; Fantacuzzi, M.; Gambacorta, N.; De Filippis, B.; Giampietro, L.; Maccallini, C.; Nicolotti, O.; Cataldi, A.; Amoroso, R. Design, synthesis and biological evaluation of imidazole and triazole-based carbamates as novel aromatase inhibitors. Eur. J. Med. Chem. 2021, 211, 113115.
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70.
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming endocrine resistance in breast cancer. Cancer Cell 2020, 37, 496–513.
- Kalra, S.; Joshi, G.; Munshi, A.; Kumar, R. Structural insights of cyclin dependent kinases: Implications in design of selective inhibitors. Eur. J. Med. Chem. 2017, 142, 424–458.
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39.
- Scheiblecker, L.; Kollmann, K.; Sexl, V. CDK4/6 and MAPK-crosstalk as opportunity for cancer treatment. Pharmaceuticals 2020, 13, 418.
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122.
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276.
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schafer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.I.; et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 2013, 24, 167–181.
- Roovers, K.; Assoian, R.K. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays 2000, 22, 818–826.
- Xiong, Y.; Li, T.; Assani, G.; Ling, H.; Zhou, Q.; Zeng, Y.; Zhou, F.; Zhou, Y. Ribociclib, a selective cyclin D kinase 4/6 inhibitor, inhibits proliferation and induces apoptosis of human cervical cancer in vitro and in vivo. Biomed. Pharmacother. 2019, 112, 108602–108613.
- Roskoski, R., Jr. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol. Res. 2016, 107, 249–275.
- Lynce, F.; Shajahan-Haq, A.N.; Swain, S.M. CDK4/6 inhibitors in breast cancer therapy: Current practice and future opportunities. Pharmacol. Ther. 2018, 191, 65–73.
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to peptide inhibitors. Cancers 2015, 7, 179–237.
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146.
- Pernas, S.; Tolaney, S.M.; Winer, E.P.; Goel, S. CDK4/6 inhibition in breast cancer: Current practice and future directions. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786451.
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 inhibition in cancer: Beyond cell cycle arrest. Trends Cell Biol. 2018, 28, 911–925.
- Dos Santos Paparidis, N.F.; Canduri, F. The emerging picture of CDK11: Genetic, functional and medicinal aspects. Curr. Med. Chem. 2018, 25, 880–888.
- Shazzad Hossain Prince, G.M.; Yang, T.-Y.; Lin, H.; Chen, M.-C. Mechanistic insight of cyclin-dependent kinase 5 in modulating lung cancer growth. Chin. J. Physiol. 2019, 62, 231–240.
- Pozo, K.; Bibb, J.A. The emerging role of Cdk5 in cancer. Trends Cancer 2016, 2, 606–618.
- Xi, M.; Chen, T.; Wu, C.; Gao, Z.; Wu, Y.; Luo, X.; Du, K.; Yu, L.; Cai, T.; Shen, R.; et al. CDK8 as a therapeutic target for cancers and recent developments in discovery of CDK8 inhibitors. Eur. J. Med. Chem. 2019, 164, 77–91.
- Eyvazis, S.; Hejazi, M.S.; Kahora, H.; Abasi, M.; Zamiri, R.E.; Tarhriz, V. CDK9 as an appealing target for therapeutic interventions. Curr. Drug Targets 2019, 20, 453–464.
- Bose, P.; Simmons, G.L.; Grant, S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin. Investig. Drugs 2013, 22, 723–738.
- Blum, K.A.; Ruppert, A.S.; Woyach, J.A.; Jones, J.A.; Andritsos, L.; Flynn, J.M.; Rovin, B.; Villalona-Calero, M.; Ji, J.; Phelps, M.; et al. Risk factors for tumor lysis syndrome in patients with chronic lymphocytic leukemia treated with the cyclin-dependent kinase inhibitor, flavopiridol. Leukemia 2011, 25, 1444–1451.
- Le Tourneau, C.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250.
- Payton, M.; Chung, G.; Yakowec, P.; Wong, A.; Powers, D.; Xiong, L.; Zhang, N.; Leal, J.; Bush, T.L.; Santora, V.; et al. Discovery and evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res. 2006, 66, 4299–4308.
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353.
- Teng, Y.; Lu, K.; Zhang, Q.; Zhao, L.; Huang, Y.; Ingarra, A.M.; Galons, H.; Li, T.; Cui, S.; Yu, P.; et al. Recent advances in the development of cyclin-dependent kinase 7 inhibitors. Eur. J. Med. Chem. 2019, 183, 1116412.
- Czudor, Z.; Balogh, M.; Bánhegyi, P.; Boros, S.; Breza, N.; Dobos, J.; Fábián, M.; Horváth, Z.; Illyés, E.; Markó, P.; et al. Novel compounds with potent CDK9 inhibitory activity for the treatment of myeloma. Bioorg. Med. Chem. Lett. 2018, 28, 769–773.
- Dukelow, T.; Kishan, D.; Khasraw, M.; Murphy, C.G. CDK4/6 inhibitors in breast cancer. Anticancer Drugs 2015, 26, 797–806.
- Poratti, M.; Marzaro, G. Third-generation CDK inhibitors: A review on the synthesis and binding modes of palbociclib, ribociclib and abemaciclib. Eur. J. Med. Chem. 2019, 172, 143–153.
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA approval: Palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin. Cancer Res. 2015, 21, 4760–4766.
- Syed, Y.Y. Ribociclib: First global approval. Drugs 2017, 77, 799–807.
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753.
- Tan, A.R.; Wright, G.S.; Thummala, A.R.; Danso, M.A.; Popovic, L.; Pluard, T.J.; Han, H.S.; Vojnović, Ž.; Vasev, N.; Ma, L.; et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: A multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 2019, 20, 1587–1601.
- Hart, L.L.; Ferrarotto, R.; Andric, Z.G.; Beck, J.T.; Subramanian, J.T.; Radosavljevic, D.Z.; Zaric, B.; Hanna, W.T.; Aljumaily, R.; Owonikoko, T.K.; et al. Myelopreservation with trilaciclib in patients receiving topotecan for small cell lung cancer: Results from a randomized, double-blind, placebo-controlled phase II study. Adv. Ther. 2021, 38, 350–365.
- Bisi, J.E.; Sorrentino, J.A.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical characterization of G1T28: A novel CDK4/6 inhibitor for reduction of chemotherapy-induced myelosuppression. Mol. Cancer Ther. 2016, 15, 783–793.
- He, S.; Roberts, P.J.; Sorrentino, J.A.; Bisi, J.E.; Storrie-White, H.; Tiessen, R.G.; Makhuli, K.M.; Wargin, W.A.; Tadema, H.; van Hoogdalem, E.J.; et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci. Transl. Med. 2017, 9, eaal3986.
- Bisi, J.E.; Sorrentino, J.A.; Jordan, J.L.; Darr, D.D.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget 2017, 8, 42343–42358.
- Stice, J.P.; Wardell, S.E.; Norris, J.D.; Yllanes, A.P.; Alley, H.M.; Haney, V.O.; White, H.S.; Safi, R.; Winter, P.S.; Cocce, K.J.; et al. CDK4/6 therapeutic intervention and viable alternative to taxanes in CRPC. Mol. Cancer Res. 2017, 15, 660–669.
- Long, F.; He, Y.; Fu, H.; Li, Y.; Bao, X.; Wang, Q.; Wang, Y.; Xie, C.; Lou, L. Preclinical characterization of SHR6390, a novel CDK 4/6 inhibitor, in vitro and in human tumor xenograft models. Cancer Sci. 2019, 110, 1420–1430.
- Wang, J.; Li, Q.; Yuan, J.; Wang, J.; Chen, Z.; Liu, Z.; Li, Z.; Lai, Y.; Gao, J.; Shen, L. CDK4/6 inhibitor-SHR6390 exerts potent antitumor activity in esophageal squamous cell carcinoma by inhibiting phosphorylated Rb and inducing G1 cell cycle arrest. J. Transl. Med. 2017, 15, 127.
- Wang, Y.; Wang, J.; Ding, L. Benzimidazole Derivatives, Preparation Methods and Uses Thereof. PCT. International Patent WO2016145622A1, 22 September 2016.
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837.
- Tate, S.C.; Cai, S.; Ajamie, R.T.; Burke, T.; Beckmann, R.P.; Chan, E.M.; De Dios, A.; Wishart, G.N.; Gelbert, L.M.; Cronier, D.M. Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin. Cancer Res. 2014, 20, 3763.
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013, 19, 6173.
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.A.; et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther. 2016, 15, 2273.
- Tsai, C.J.; Nussinov, R. The molecular basis of targeting protein kinases in cancer therapeutics. Semin. Cancer Biol. 2013, 23, 235–242.
- Hojjat-Farsangi, M. Small-molecule inhibitors of the receptor tyrosine kinases: Promising tools for targeted cancer therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801.
- Zhou, M.; Wang, R. Small-molecule regulators of autophagy and their potential therapeutic applications. ChemMedChem 2013, 8, 694–707.
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23.
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50.
- Meisel, J.E.; Chang, M. Selective small-molecule inhibitors as chemical tools to define the roles of matrix metalloproteinases in disease. BBA Mol. Cell Res. 2017, 1864, 2001–2014.
- Tadesse, S.; Yu, M.; Mekonnen, L.B.; Lam, F.; Islam, S.; Tomusange, K.; Rahaman, M.H.; Noll, B.; Basnet, S.K.C.; Teo, T.; et al. Highly potent, selective, and orally bioavailable 4-thiazol N-(pyridin-2-yl)pyrimidin-2-amine cyclin-dependent kinases 4 and 6 inhibitors as anticancer drug candidates: Design, synthesis, and evaluation. J. Med. Chem. 2017, 60, 1892–1915.
- Shao, H.; Shi, S.; Huang, S.; Hole, A.J.; Abbas, A.Y.; Baumli, S.; Liu, X.; Lam, F.; Foley, D.W.; Fischer, P.M.; et al. Substituted 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines are highly active CDK9 inhibitors: Synthesis, X-ray crystal structures, structure-activity relationship, and anticancer activities. J. Med. Chem. 2013, 56, 640–659.
- Tadesse, S.; Zhu, G.; Mekonnen, L.B.; Lenjisa, J.L.; Yu, M.; Brown, M.P.; Wang, S. A novel series of N-(pyridin-2-yl)-4-(thiazol5-yl)pyrimidin-2-amines as highly potent CDK4/6 inhibitors. Future Med. Chem. 2017, 9, 1495–1506.
- Tadesse, S.; Bantie, L.; Tomusange, K.; Yu, M.; Islam, S.; Bykovska, N.; Noll, B.; Zhu, G.; Li, P.; Lam, F.; et al. Discovery and pharmacological characterization of a novel series of highly selective inhibitors of cyclin-dependent kinases 4 and 6 as anticancer agents. Br. J. Pharmacol. 2018, 175, 2399–2413.
- Zha, C.; Deng, W.; Fu, Y.; Tang, S.; Lan, X.; Ye, Y.; Su, Y.; Jiang, L.; Chen, Y.; Huang, Y.; et al. Design, synthesis and biological evaluation of tetrahydronaphthyridine derivatives as bioavailable CDK4/6 inhibitors for cancer therapy. Eur. J. Med. Chem. 2018, 148, 140–153.
- Fu, Y.; Tang, S.; Su, Y.; Lan, X.; Ye, Y.; Zha, C.; Li, L.; Cao, J.; Chen, Y.; Jiang, L.; et al. Discovery of a class of diheteroaromatic amines as orally bioavailable CDK1/4/6 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5332–5336.
- Wang, Y.; Liu, W.-J.; Yin, L.; Li, H.; Chen, Z.-H.; Zhu, D.-X.; Song, X.-Q.; Cheng, Z.-Z.; Song, P.; Wang, Z.; et al. Design and synthesis of 4-(2,3-dihydro-1H-benzo[d]pyrrolo[1,2-a] imidazol-7-yl)-N-(5-(piperazin-1-ylmethyl) pyridine-2-yl)pyrimidin-2-amine as a highly potent and selective cyclin-dependent kinases 4 and 6 inhibitors and the discovery of structure-activity relationships. Bioorg. Med. Chem. Lett. 2018, 28, 974–978.
- Shi, C.; Wang, Q.; Liao, X.; Ge, H.; Huo, G.; Zhang, L.; Chen, N.; Zhai, X.; Hong, Y.; Wang, L.; et al. Discovery of 6-(2-(dimethylamino)ethyl)-N-(5-fluoro-4-(4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole-6-yl)pyrimidin-2-yl)-5,6,7,8-tetrahydro-1,6-naphthyridin-2-amine as a highly potent cyclin-dependent kinase 4/6 inhibitor for treatment of cancer. Eur. J. Med. Chem. 2019, 178, 352–364.
- Abbas, S.E.-S.; George, R.F.; Samir, E.M.; Aref, M.M.A.; Abdel-Aziz, H.A. Synthesis and anticancer activity of some pyrido[2,3-d]pyrimidine derivatives as apoptosis inducers and cyclin-dependent kinase inhibitors. Future Med. Chem. 2019, 11, 2395–2414.
- Shi, C.; Wang, Q.; Liao, X.; Ge, H.; Huo, G.; Zhang, L.; Chen, N.; Zhai, X.; Hong, Y.; Wang, L.; et al. Discovery of a novel series of imidazo[10,2′:1,6]pyrido[2,3-d]pyrimidin derivatives as potent cyclin-dependent kinase 4/6 inhibitors. Eur. J. Med. Chem. 2020, 193, 112239.
- Zhao, H.; Hu, X.; Cao, K.; Zhang, Y.; Zhao, K.; Tang, C.; Feng, B. Synthesis and SAR of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as potent and selective CDK4/6 inhibitors. Eur. J. Med. Chem. 2018, 157, 935–945.
- Garg, M.; Chauhan, M.; Singh, P.K.; Alex, J.M.; Kumar, R. Pyrazoloquinazolines: Synthetic strategies and bioactivities. Eur. J. Med. Chem. 2015, 97, 444–461.
- Toure, M.; Crews, C.M. Small-molecule PROTACS: New approaches to protein degradation. Angew. Chem. Int. Ed. Engl. 2016, 55, 1966–1973.
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114.
- Marak, B.N.; Dowarah, J.; Khiangte, L.; Singh, V.P. A comprehensive insight on the recent development of cyclic dependent kinase inhibitors as anticancer agents. Eur. J. Med. Chem. 2020, 203, 112571.
- Crews, C.M. Targeting the undruggable proteome: The small molecules of my dreams. Chem. Biol. 2010, 17, 551–555.
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50.
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.-H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521.
- Gao, H.; Sun, X.; Rao, Y. PROTAC technology: Opportunities and challenges. ACS Med. Chem. Lett. 2020, 11, 237–240.
- Robb, C.M.; Contreras, J.I.; Kour, S.; Taylor, M.A.; Abid, M.; Sonawane, Y.A.; Zahid, M.; Murry, D.J.; Natarajan, A.; Rana, S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580.
- Hatcher, J.M.; Wang, E.S.; Johannessen, L.; Kwiatkowski, N.; Sim, T.; Gray, N.S. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med. Chem. Lett. 2018, 9, 540–545.
- Zhao, B.; Burgess, K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem. Commun. 2019, 55, 2704–2707.
- Rana, S.; Bendjennat, M.; Kour, S.; King, H.M.; Kizhake, S.; Zahid, M.; Natarajan, A. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 2019, 29, 1375–1379.
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J. Med. Chem. 2019, 62, 7575–7582.
- Jiang, B.; Wang, E.S.; Donovan, K.A.; Liang, Y.; Fischer, E.S.; Zhang, T.; Gray, N.S. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew. Chem. Int. Ed. 2019, 58, 6321–6326.
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309.
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305.
- Huang, T.; Dobrovolsky, D.; Paulk, J.; Yang, G.; Weisberg, E.L.; Doctor, Z.M.; Buckley, D.L.; Cho, J.H.; Ko, E.; Jang, J.; et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 2018, 25, 88–99.e6.
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.
- Anderson, N.A.; Cryan, J.; Ahmed, A.; Dai, H.; McGonagle, G.A.; Rozier, C.; Benowitz, A.B. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg. Med. Chem. Lett. 2020, 30, 127106.
- Steinebach, C.; Ng, Y.L.D.; Sosič, I.; Lee, C.-S.; Chen, S.; Lindner, S.; Vu, L.P.; Bricelj, A.; Haschemi, R.; Monschke, M.; et al. Systematic exploration of different E3 ubiquitin ligases: An approach towards potent and selective CDK6 degraders. Chem. Sci. 2020, 11, 3474–3486.

