 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Walter Manucha | + 1893 word(s) | 1893 | 2021-04-22 06:26:59 | | | |
| 2 | Nora Tang | Meta information modification | 1893 | 2021-04-27 05:23:49 | | |
Video Upload Options
Exaggerated oxidative stress and hyper-inflammation are essential features of oxidative/inflammatory diseases. Simultaneously, both processes may be the cause or consequence of mitochondrial dysfunction, thus establishing a vicious cycle among these three factors. However, several natural substances, including melatonin and micronutrients, may prevent or attenuate mitochondrial damage and may preserve an optimal state of health by managing the general oxidative and inflammatory status.
1. Introduction
Mitochondrial dysfunction is associated with impaired immune and inflammatory responses. It has been suggested that inflammation provoked by mitochondrial dysfunction is responsible for the explosive release of proinflammatory cytokines [1], contributing to multiple oxidative/inflammatory diseases, such as sepsis, neurodegenerative pathologies, inflammatory bowel disease, cardiovascular and metabolic disorders, and respiratory diseases, including complications and death by COVID-19 as one of the most relevant current examples. The systemic hyper-inflammation usually observed in these pathologies is known as cytokine storm or macrophage activation syndrome [2].
Notably, the preservation of mitochondrial health through healthy life habits, pharmacological agents, or nutritional supplements provides significantly improved mitochondrial dynamic and activity, reducing inflammation and oxidation stress [1].
In this regard, it is known that melatonin and micronutrients are essential elements that are required by living beings including humans in order to perform multiple metabolic and physiological functions for maintaining an optimal state of health [3][4]. Interestingly, both melatonin and many of these micronutrients exert several of their actions at the mitochondrial level [5].
2. Mitochondrion: Function, Dynamics, and Dysfunction
Mitochondria are cellular organelles that perform numerous essential functions that ensure homeostasis. They play a crucial role in energy metabolism by synthesizing adenosine triphosphate (ATP) through oxidative phosphorylation. Both glucose and fatty acids metabolites enter the tricarboxylic acid (TCA) cycle to produce ATP [6]. Mitochondria also participate in the regulation of cell calcium homeostasis, apoptosis, and they are an important source of reactive oxygen species (ROS). Mitochondrial ATP generation depends on the transfer of electrons along the transport chain (ETC), which is coupled to proton transport across the inner mitochondrial membrane, establishing an electrochemical gradient that is capable of driving ATP synthesis. Mitochondria are dynamic organelles that change their morphology in response to physiological and pathological stimuli, which also affect their functions. Mitochondrial dynamics comprises several processes: mitochondrial biogenesis, fusion, fission, and mitophagy [7]. A number of pathological situations including cardiovascular, metabolic, and inflammatory diseases are associated with mitochondrial dysfunction. Aberrant mitochondrial physiology appears to be the result of energetics and/or dynamics alterations. Disturbances of mitochondrial ETC not only lead to reduced ATP production but to modifications of mitochondrial morphology [8]. In turn, an imbalance in mitochondrial dynamics decreases the efficiency of mitochondrial energy production [6][9]. Finally, mitochondrial dysfunction is considered the main source of ROS in cells, and it contributes to the development and progression of many pathologies, including inflammation (Figure 1) [10].
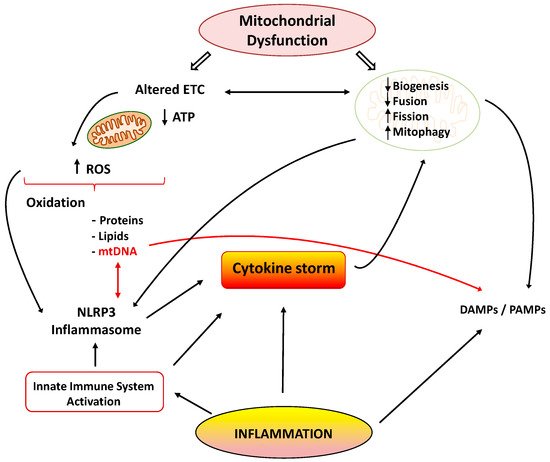
Figure 1. Cytokine storm: link between inflammation and mitochondrial dysfunction. ETC: electrons transport chain; ROS: reactive oxygen species; NLRP3: NOD-, LRR- and pyrin domain-containing 3; DAMPS/PAMPS: damage/pathogen-associated molecular patterns.
3. Inflammation and Mitochondrial Dysfunction
Inflammation is a complex, protective response of the body to infections and tissue damage. The inflammatory response includes activation of the immune system to repair damaged tissue and defend against pathogens through the secretion of specific mediators. However, when inflammation persists, it produces tissue damage in many diseases.
Many reports have shown the association between the inflammatory process and mitochondrial dysfunction. Inflammation promotes mitochondrial dysfunction, and dysfunctional mitochondrion participates in the pathogenesis of inflammation via several mechanisms, which may establish a vicious circle of recurrent inflammation [11]. The development and progression of several inflammatory disorders are accompanied by mitochondrial dysfunction and enhanced ROS production [12]. Both acute and chronic inflammatory diseases are characterized by the exaggerated generation of oxygen-based reactive species, which produce damage in mitochondrial proteins, lipids, and mitochondrial DNA (mtDNA) (Figure 1). These alterations negatively influence normal mitochondrial function and dynamics [13]. Inducible nitric oxide synthase (iNOS) activity is also elevated in the mitochondria during inflammation, leading to enhanced mitochondrial NO production and reactive nitrogen species (RNS). Both ROS and RNS reduce respiratory chain activity and ATP production, produce mtDNA alterations, and finally lead to cell damage and death [14][15].
3.1. Inflammation Alters Mitochondrial Energetics
Inflammatory mediators can also alter activities of the TCA cycle. TNF-α and IL-1 reduce pyruvate dehydrogenase (PDH) activity, together with a concomitant reduction of complex I and II activities [16]. A reduction of PDH activity by inflammatory cytokines has been shown in several cell types such as cardiomyocites, hepatic and skeletal muscle cells, and in cells exposed to septic stimuli [17]. Reduced PDH activity worsens mitochondrial dysfunction, because less acetyl-coenzyme A is produced. Furthermore, this situation reduces the ability of mitochondria to produce melatonin intrinsically, and consequently, it also worsens inflammatory processes [18][19]. Melatonin is a powerful anti-inflammatory agent, and its loss at the mitochondrial level would certainly exaggerate the inflammatory response. Alpha-ketoglutarate dehydrogenase (KGDH) is a rate-limiting enzyme in the TCA cycle, and it is a key element for the mitochondrial ETC [20]. Reduced KGDH activity has been observed during inflammatory conditions including inflamed neural tissue in Alzheimer’s disease [21].
During oxidative phosphorylation (OXPHOS), NADH generated by the TCA cycle is oxidized and provides electrons to the ETC. A reduced expression of genes encoding subunits of complexes I to IV and ATP synthase has been observed in several pathological inflammatory situations. Acute systemic inflammation induced by intravenous lipopolysaccharide (LPS) administration exerts inhibitory effects on OXPHOS [22]. ETC complexes I, II, and IV mRNA and protein levels were down-regulated in muscle and liver cells incubated with LPS [23]. Systemic LPS administration in rodents leads to a TLR4-mediated burst of pro-inflammatory cytokines, which alter mitochondrial energy production. Recent studies showed that TNF-α was able to reduce activity of complex I, III, and IV, leading to decreased ATP production [24]. TNF-α also alters mitochondrial biogenesis regulation in various cell types and tissues through the reduction of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) expression [25].
3.2. Inflammation Alters Mitochondrial Dynamics and Cell Death Pathways
There are many studies of the deleterious effects of inflammatory agents on mitochondrial dynamics. It has been reported that TNF-α induces mitochondrial dysfunction and altered morphology with small condensed mitochondria in adipocytes [26]. This effect may be related to the increased expression of fission protein Fis1 and the reduced expression of fusion protein Opa1. Decreased expression of Opa1 also led to mitochondrial fragmentation [27]. Moreover, IL1-β was able to induce mitochondrial fragmentation and respiration impairment through the fission protein Drp1 in astrocytes. IL-6 is also involved in the lowered expression of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) and fusion proteins Mfn1 and 2 during the initial stages of cachexia [28]. Finally, other reports also support the interplay between inflammation and mitophagy, leading to apoptosis and cell death (Figure 1) [29].
3.3. Mitochondrial Dysfunction Promotes Inflammation
As previously mentioned, inflammation and mitochondrial dysfunction have mutually destructive actions and induce a vicious cycle of function deterioration. Structural and functional alterations of mitochondria can stimulate the production of inflammatory mediators, which in turn can further impair mitochondrial function. It has been shown that ROS are able to promote inflammation through NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome [30]. Furthermore, mitochondria are considered the main activators of the NLRP3 inflammasome, and they play a crucial role in the control of innate immunity and in the inflammatory response. In fact, interrelationships between mtDNA and NLRP3-inflammasome activation support the involvement of the innate immune pathways in disease coursing with mitochondrial dysfunction [31]. The NLRP3 inflammasome acts as a sensor of mitochondrial dysfunction. Activation of this complex leads to the production of IL1β, which in turn causes loss of mitochondrial membrane potential, reduction of ATP levels, and ROS generation. These findings support the notion that cytokines can produce mitochondrial dysfunction, leading to a vicious degenerative cycle [11]. Another mechanism by which dysfunctional mitochondria evoke inflammatory responses involves the release of damage-associated molecular patterns (DAMPs) into the cytoplasm. DAMPs are macromolecules that are able to induce local inflammatory responses during infections or stress [32]. Due to similarities to bacterial DNA, altered mtDNA can be considered as a DAMP. Moreover, mtDNA is involved in the innate immune response, and thus, specific inflammatory pathologies are directly related to mtDNA alterations [33]. Under normal conditions, defective mitochondrial mtDNA are degraded and eliminated by mitophagy. However, when mitochondrial damage is elevated, mtDNA cannot be effectively eliminated, and it can activate NLRP3 inflammasome-dependent pathways [34].
Activation the TLR-9 pathway is another mechanism by which mtDNA exerts inflammatory responses with the subsequent generation of nitric oxide and TNF-α [35]. It is important to note that normal mitochondria in the presence of TNF-α activate macrophages and cytokine production. Thus, it could be concluded that damaged mitochondria elicit exaggerated inflammatory responses through ROS production and the release of DAMPs, leading to a deleterious vicious cycle (Figure 1).
4. Relevance of Mitochondrial Dysfunction in the Pathogenesis of Inflammation in Sepsis and COVID-19
Sepsis is a generalized state of inflammation with an initial acute hyperinflammatory phase as a response to infection followed by a hypoinflammatory phase, which is immune-tolerant [36]. Several metabolic processes are re-programmed in each phase, and they play differential roles in the pathophysiological manifestations of the two phases. The hyperinflammatory phase is characterized by increased aerobic glycolysis capacity and oxygen consumption for ETC in many cell types, including monocytes. Activation of the respiratory rate and energy production are essential for the promotion of cytokine production and phagocytosis, which are key elements in host defense mechanisms [37]. This initial phase consists in the reaction of host tissues to enhance energy production in order to increase pathogen-killing capacity of the innate immune cells and attempt to control the spread of the infection [38].
The late hypoinflammatory, immune-tolerant phase is characterized by increased OXPHOS in immune cells [39]. During the second phase of sepsis, mitochondrial respiration and ATP production are partially restored. This hypometabolic state is cytoprotective and immunosuppressive, but it actually impairs recovery and infection control [38]. This phase is associated with a decrease in the cytokine storm and with restoration of sirtuin activity. Sirtuins type 3, 4, and 5 are localized in the mitochondria and are known for their anti-inflammatory and anti-oxidant properties. Increased sirtuin activity promotes OXPHOS and reduces glycolysis [38][39]. Thus, the mitochondrial biogenesis mediators PGC-1α, mitochondrial transcription factor A (TFAM), and nuclear respiratory factor 1 (NRF-1) are upregulated in the second phase of sepsis [40]. As previously mentioned, the release of large amounts of cytokines and chemokines by immune cells produces a sustained systemic inflammatory response that causes the acute respiratory distress syndrome in COVID-19 patients. Indeed, the patients with severe respiratory infection present higher levels of the cytokines than patients with less severe symptoms [41]. Mitochondria seem to be involved in the cytokine storm caused by SARS-CoV-2. A recent analysis of gene expression in the SARS-CoV-2 infected lung cell lines demonstrated an upregulation of genes involved in mitochondrial cytokine signaling and downregulation in the mitochondrial organization, respiration, and autophagy genes [42]. Finally, during the hyperinflammatory phase of COVID-19-related sepsis, immune cells adapt their metabolism, favoring glycolysis over OXPHOS for ATP production. These metabolic changes enhance macrophage phagocytic action and the further synthesis of cytokines and chemokines in a kind of self-perpetuating cycle [43]. Collectively, all these findings provide evidence that mitochondrial dysfunction impairs immune response as well as increases inflammation and severity in the COVID-19-related sepsis. It should be mentioned that OXPHOS and TCA cycle inhibition in mitochondria reduce the synthesis of certain molecules, including melatonin. This supports the use and benefits of melatonin as potential adjuvant treatment strategy to reduce the severity of the COVID-19 [43]. This aspect will be discussed later in this report.
References
- Fernández-Ayala, D.J.M.; Navas, P.; López-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147.
- Bektas, A.; Schurman, S.H.; Franceschi, C.; Ferrucci, L. A public health perspective of aging: Do hyper-inflammatory syndromes such as COVID-19, SARS, ARDS, cytokine storm syndrome, and post-ICU syndrome accelerate short- and long-term inflammaging? Immun. Ageing 2020, 17, 1–10.
- Maggini, S.; Pierre, A.; Calder, P.C. Immune Function and Micronutrient Requirements Change over the Life Course. Nutrients 2018, 10, 1531.
- Shenkin, A. Micronutrients in health and disease. Postgrad. Med. J. 2006, 82, 559–567.
- Wesselink, E.; Koekkoek, W.; Grefte, S.; Witkamp, R.; van Zanten, A. Feeding mitochondria: Potential role of nutritional components to improve critical illness convalescence. Clin. Nutr. 2019, 38, 982–995.
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 418–426.
- Lahera, V.; de Las Heras, N.; López-Farré, A.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11.
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848.
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 1833–1838.
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569.
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931.
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, Á.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, Oxidative Stress, and Obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132.
- Crimi, E.; Sica, V.; Slutsky, A.S.; Zhang, H.; Williams-Ignarro, S.; Ignarro, L.J.; Napoli, C. Role of oxidative stress in experimental sepsis and multisystem organ dysfunction. Free. Radic. Res. 2006, 40, 665–672.
- Bartz, R.R.; Suliman, H.B.; Fu, P.; Welty-Wolf, K.; Carraway, M.S.; MacGarvey, N.C.; Withers, C.M.; Sweeney, T.E.; Piantadosi, C.A. Staphylococcus aureusSepsis and Mitochondrial Accrual of the 8-Oxoguanine DNA Glycosylase DNA Repair Enzyme in Mice. Am. J. Respir. Crit. Care Med. 2011, 183, 226–233.
- Escames, G.; López, L.C.; Ortiz, F.; López, A.; García, J.A.; Ros, E.; Acuña-Castroviejo, D. Attenuation of cardiac mitochondrial dysfunction by melatonin in septic mice. FEBS J. 2007, 274, 2135–2147.
- Zell, R.; Geck, P.; Werdan, K.; Boekstegers, P. Tnf-α and IL-1α inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: Evidence for primary impairment of mitochondrial function. Mol. Cell. Biochem. 1997, 177, 61–67.
- Vary, T.C.; Hazen, S. Sepsis alters pyruvate dehydrogenase kinase activity in skeletal muscle. Mol. Cell. Biochem. 1999, 198, 113–118.
- Reiter, R.J.; Ma, Q.; Sharma, R. Melatonin in Mitochondria: Mitigating Clear and Present Dangers. Physiology 2020, 35, 86–95.
- Reiter, R.J.; Sharma, R.; de Campos Zuccari, D.A.P.; de Almeida Chuffa, L.G.; Manucha, W.; Rodriguez, C. Melatonin synthesis in and uptake by mitochondria: Implications for diseased cells with dysfunctional mitochondria. Futur. Med. Chem. 2021, 13, 335–339.
- Sheu, K.-F.R.; Blass, J.P. The alpha-Ketoglutarate Dehydrogenase Complex. Ann. N. Y. Acad. Sci. 1999, 893, 61–78.
- Mastrogiacomo, F.; Bergeron, C.; Kish, S.J. Brain α-Ketoglutarate Dehydrotenase Complex Activity in Alzheimer’s Disease. J. Neurochem. 1993, 61, 2007–2014.
- Lee, I.; Hüttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1579–1586.
- Suliman, H.B.; Welty-Wolf, K.E.; Carraway, M.; Tatro, L.; Piantadosi, C.A. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 2004, 64, 279–288.
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-α regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 451–461.
- Palomer, X.; Álvarez-Guardia, D.; Rodríguez-Calvo, R.; Coll, T.; Laguna, J.C.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Vázquez-Carrera, M. TNF-α reduces PGC-1α expression through NF-κB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc. Res. 2008, 81, 703–712.
- Chen, X.-H.; Zhao, Y.-P.; Xue, M.; Ji, C.-B.; Gao, C.-L.; Zhu, J.-G.; Qin, D.-N.; Kou, C.-Z.; Qin, X.-H.; Tong, M.-L.; et al. TNF-α induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 328, 63–69.
- Hahn, W.S.; Kuzmicic, J.; Burrill, J.S.; Donoghue, M.A.; Foncea, R.; Jensen, M.D.; Lavandero, S.; Arriaga, E.A.; Bernlohr, D.A. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Metab. 2014, 306, E1033–E1045.
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14.
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147.
- Moro, L. Mitochondria at the Crossroads of Physiology and Pathology. J. Clin. Med. 2020, 9, 1971.
- DiMauro, S.; Schon, E.A. Mitochondrial Disorders in the Nervous System. Annu. Rev. Neurosci. 2008, 31, 91–123.
- Cardaci, S.; Ciriolo, M.R. TCA Cycle Defects and Cancer: When Metabolism Tunes Redox State. Int. J. Cell Biol. 2012, 2012, 161837.
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230.
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664.
- Arbini, A.A.; Guerra, F.; Greco, M.; Marra, E.; Gandee, L.; Xiao, G.; Lotan, Y.; Gasparre, G.; Hsieh, J.-T.; Moro, L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis 2013, 2, e82.
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1–9.
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human Monocytes Undergo Functional Re-programming during Sepsis Mediated by Hypoxia-Inducible Factor-1α. Immunity 2015, 42, 484–498.
- Wang, X.; Buechler, N.L.; Woodruff, A.G.; Long, D.L.; Zabalawi, M.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Sirtuins and Immuno-Metabolism of Sepsis. Int. J. Mol. Sci. 2018, 19, 2738.
- Vachharajani, V.; McCall, C.E. Sirtuins: Potential therapeutic targets for regulating acute inflammatory response? Expert Opin. Ther. Targets 2020, 24, 489–497.
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981.
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32.
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209.
- Reiter, R.J.; Sharma, R.; Ma, Q.; Dominquez-Rodriguez, A.; Marik, P.E.; Abreu-Gonzalez, P. Melatonin Inhibits COVID-19-induced Cytokine Storm by Reversing Aerobic Glycolysis in Immune Cells: A Mechanistic Analysis. Med. Drug Discov. 2020, 6, 100044.


