 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Serena Pillozzi | + 2094 word(s) | 2094 | 2021-04-22 05:18:43 | | | |
| 2 | Dean Liu | -11 word(s) | 2083 | 2021-04-22 09:36:02 | | | | |
| 3 | Dean Liu | Meta information modification | 2083 | 2021-04-29 04:24:03 | | |
Video Upload Options
RET-selective tyrosine kinase inhibitors (TKIs) pralsetinib and selpercatinib, are effective against the RET V804L/M gatekeeper mutants. However, adaptive mutations that cause resistance at the solvent front RET G810 residue have been found, pointing to the need for the development of the next-generation of RET-specific TKIs.
1. Introduction
Recent evidence in non-small cell lung cancer (NSCLC) about the new highly selective REarranged during Transfection (RET) inhibitors selpercatinib and pralsetinib, despite impressive results in clinical trials, have raised the urgency to highlight the best therapeutic sequence in view of novel resistances which are able to cause selective inhibition to be useless. With this aim, and aware that the inhibition of RET has always been a challenge since its discovery in T-cell lymphoma [1], we want to summarize the available knowledge on RET inhibitors, including both ongoing trials with new drugs and pre-clinical data concerning overcoming the resistance mechanisms.
2. RET in Pills
RET is located on chromosome 10q11.2 and its expression is mediated by several DNA-binding proteins belonging to the Sp family of transcription factors (Sp1, Sp3) [2] or early growth response protein 1 (EGR1) [2], SRY-box 10 (SOX10), paired box 3 (PAX3) [3], NK2 homeobox 1 (NKX2–1) and homeobox B5 (HOXB5) [4]. RET encodes for a Transmembrane Tyrosine Kinase Receptor (RTK) with a unique structure composed of four cadherin-like domains, a cysteine-rich domain, a transmembrane domain and a tyrosine kinase (TK) domain, this latter has a different number of amino acids depending on the isoform transcribed (RET9, RET43 and RET51) [5]. Each isoform interacts with adaptors and signaling proteins that are able to activate different downstream pathways during embryogenesis, in homeostasis of several tissues [6]. Physiologically, beginning RET signals depend on the binding of specific ligand members of the glial cell line-derived neurotrophic factors (GDNFs) with GDNF family receptor alpha (GFRα). The ligand family includes GDNF, neurturin (NTRN), artemin (ARTN) and persephin (PSPN) and each has a selective, although not completely specific, receptor, respectively called GFRα1, GFRα2, GFRα3 and GFRα4 [7]. The interposition of the GDNF-GFRα complex allows for the homodimerization between RET monomers resulting in autophosphorylation of the intracellular tyrosine residues of the main docking-site of the RET51 isoform (Y1062). RET is also able to heterodimerize with other RTKs [5]. Phosphorylated tyrosine recruits a multitude of adaptors that, in turn, mediate the activation of RAS- Mitogen-Activated Protein Kinases (MAPK) and Phosphatidylinositol-3 Kinase (PI3K)- Protein Kinase B (AKT) pathways [5]. Several docking sites (Y900, Y905, Y981, Y1015 and Y1096), able to trigger additional downstream pathways such as JAK/STAT, PKA, PKC and JNK, have been described [6]. Moreover, RET interacts with RTKs and other cell surface proteins guaranteeing a continuity and spreading of downstream signals [8] (Figure 1). During embryogenesis RET is mainly expressed in the urinary tract, nervous system and hematopoietic stem cells, justifying the pathogenesis of hereditary diseases secondary to germline mutations (loss of function). In adult life, low levels of RET expression are registered in all tissues [9] and different RET molecular alterations have been reported in tumors at either germline or somatic levels. These include gene amplification, fusion, as well as single base substitutions/small insertions/deletions.
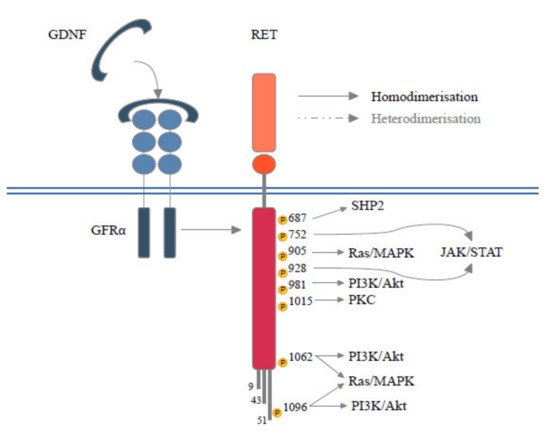
Figure 1. Schematic RET protein structure showing phosphorylation sites. RET forms a heterocomplex with GFRα and GFLs proteins, which in turn results in the activation of multiple signaling pathways involved in survival, differentiation, motility, proliferation, and growth.
2.1. Germline Mutations
Genitourinary and nervous system development [10][11], maturation and migration of stem cell lines and a general involvement in embryogenesis and spermatogenesis, represent the main known mechanisms in which RET’s signaling is involved during embryonic development [12][13]. It’s clearly understandable how RET loss of function due to germline mutations, affecting those mechanism, can lead to a variety of congenital malformations such as Hirschsprung disease (HSCR) and congenital abnormalities of the kidney and urinary tract (CAKUT), and cause numerous symptoms in patients with phenotypic variants of these syndromes [14][15]. However, a role for RET in maintenance of hematopoietic system and in development of Gut-Associated Lymphoid Tissue (GALT) has recently been recognized [16]. Germinal mutations of the proto-oncogene RET affecting cysteine-rich extracellular domains or less frequently on the intracellular domains give rise to multiple neuroendocrine neoplasia 2 (MEN2). MEN2 is classified based on clinical features in MEN2A characterized by thyroid cancer, pheochromocytoma, and hyperparathyroidism and in MEN2B with also ganglioneuromatosis and a Marfanoid habitus [17]. Similarly to MEN2, the familial medullary thyroid carcinoma (FMTC) derived from germinal point mutation that causes an increase in the effect of self-activation by increasing ATP-binding or phosphorylation activity, sustains the oncogenic and pro-proliferative stimuli [18]. Every point mutation, rarely seen outside neuroendocrine neoplasms, correlates with different prognosis and clinical outcome, suggesting the necessity to sketch out an early screening and subsequently a different therapeutic approach [19][20]. Indeed, MEN2A and FMTC, having phenotypic and clinical indolent characteristics, appear to be more of a continuum of the same disease, unlike MEN2B which has a juvenile onset and a more aggressive course [21].
2.2. Somatic Mutations and Cancer
To better understand its decisive role as a proto-oncogene in sporadic cancers we had to wait until the Chernobyl disaster in 1986 showed a correlation between the papillary thyroid carcinomas (PTC) onset and gene rearrangements in post-radiation exposed children [22]. RET/Coiled-Coil Domain Containing 6 (CCDC6) gene fusion is associated in about 80% of cases of sporadic PTC, while the Nuclear Receptor Coactivator 4 (NCOA4) gene, is mainly related to radiation exposure and younger age [23]. Countless rearrangements have been described in literature being a part of the pathogenesis of PTC [24]. Although PTCs are the most frequently associated cancers. with RET rearrangements (10–20%), many other neoplasms are associated with RET-fusion involved in creating resistances and escaping mechanisms to classical therapies. In hormone positive breast cancer (BC), RET overexpression is described in less than 0.1% of cases and is involved in resistance to anti-hormonal therapies in BC cell lines [25]. Based on preclinical evidence of crosstalk between RET and positive estrogen receptors, some clinical trials in BC patients without any convincing results in disease control explore the benefit of using multi-kinase inhibitors active on RET [26]. Recently, a single case report has been presented as part of LIBRETTO-001 trial, of a metastatic BC woman who presented a complete clinical response with Selpercatinib, suggesting a possible role of selective RET inhibitors in this field [27]. In colon cancers RET rearrangements represent 0.2% of cases [28]. Among them, 2/3 manifest in the right colon and are characterized by MSI, RAS and BRAF wild type status, and could benefit from the use of specific therapies [29][30]. Other gastrointestinal malignancies, gynecological tumors, renal and prostate cancer have a limited expression of RET fusions [31] and could benefit from treatment inside basket trials, thanks to recent gene sequencing techniques.
2.3. RET in Lung Cancer
In NSCLC the prevalence of RET alterations is estimated to be 1–2% of all cases [32]. Thanks to modern genomic sequencing methods, the first fusion gene discovery in 2011 between RET and the Kinesin Family Member 5B (KIF5B) gene has allowed to broaden the knowledge of translocations involving RET [33]. As mentioned above, rearrangements involving chromosome 10 are intrachromosomal, leading to fusion with several genes lying on the same chromosome. In NSCLC the gene most involved in fusions is KIF5B, a gene involved in a pericentric rearrangement, followed by CCDC6 and NCOA4 which are characterized by a paracentric inversion fusion [32]. Several other inter-chromosomal rearrangements or translocations have been described, however they represent a small percentage of cases, we have summarized some in Table 1. [24][34]. Breakpoints in KIF5B are frequently found in the intron 11 at different positions and are involved in transcription of intracytoplasmic segments of RET, however, different introns are rarely involved in fostering the inclusion of the transmembrane dominion [35][36]. In addition to fusions, single amplifications or mutations with variable penetrance related to histotype and gender have been found [34][37]. KIF5B exon 15 fusion to RET exon 12 is the most frequently detected fusion in nonsmokers and young females, while CCDC6 exon 1 to RET exon 12 correlates with smoking habits and male gender [34].
Table 1. Other rearrangements and RET fusions.
| Fusions | Histotype | Gender | Reference |
|---|---|---|---|
| CDC123-RET | ADC | F | [42] |
| CCDC6-RET | ADC, NE | M > F | [34] |
| CLIP1-RET | ADC | NA | [43] |
| CUX1-RET | ADC | M | [44] |
| EPHA5-RET | ADC | NA | [45] |
| ERC1-RET | ADC | NA | [43] |
| FRMD4A-RET | ADC | F | [46] |
| FYCO1-RET | ADC | F | [34] |
| ITGA8-RET | ADC | M | [34] |
| ITIH2-RET | AS | F | [34] |
| KIF13A-RET | ADC | F | [47] |
| KIF5B-RET | ADC, NE, NSCLC, AS | F > M | [34] |
| KIAA1468-RET | IMA | M | [48] |
| MIR3924-RET | SCC | M | [34] |
| MYO5C-RET | ADC | NA | [49] |
| NCOA4-RET | ADC | F | [50] |
| PICALM-RET | ADC | NA | [45] |
| RASSF4-RET | ADC | NA | [51] |
| RUFY2-RET | ADC | NA | [52] |
| SLC25A36-RET | ADC | F | [34] |
| SLC39A8-RET | ADC | F | [34] |
| TBC1D32-RET | ADC | F | [53] |
| TRIM24-RET | ADC | NA | [52] |
| TRIM33-RET | ADC | F | [54] |
| WAC-RET | ADC | F | [55] |
| ZBTB41-RET | ADC | M | [34] |
Abbreviations: ADC adenocarcinoma; NE neuroendocrine; AS adenosquamous carcinoma; NSCLC non-small cell lung cancer; IMA invasive mucinous adenocarcinoma; F: female, M: male; NA not available.
3. New RET-Selective Inhibitors
None of the drugs described so far have been designed to preferentially bind to RET and, probably due to poor pharmacokinetic features and off-target side effects, were associated with modest clinical activity. It has been hypothesized that RET-specific antagonists could have achieved better clinical outcomes in patients harboring RET-rearranged NSCLC. Recently, two highly potent and selective RET TKIs, selpercatinib (LOXO-292) and pralsetinib (BLU-667), have been developed and their activity has been investigated in early phase trials. These agents, specifically tailored to target the activated forms of RET while sparing other kinases, offer the potential for a better clinical efficacy with a more satisfactory side effect profile. Selpercatinib has > 100-fold selectivity against VEGFR2 [75] and pralsetinib has 87-fold selectivity against VEGFR2 and 20-fold selectivity against JAK1 [74]. Furthermore, both are effective in inhibiting the RETV804L/M gatekeeper mutants and they are effective in the central nervous system [76]. Preclinically, selpercatinib (LOXO-292) demonstrated potent RET-selective antitumor activity both in in vitro and in vivo models, against both RET wild-type RET and RET alterations, with minimal activity against other kinase targets [75][77]. In a clinical setting, recent results from phase 1/2 LIBRETTO-001 trial [73] reported that selpercatinib achieved durable ORs in patients with advanced NSCLC marked by RET gene fusions. Of the first 105 enrolled patients with RET fusion-positive NSCLC, previously treated with platinum-based chemotherapy, the ORR was 64%, including two patients (2%) with a complete response and 65 patients (62%) with partial response. The median duration of response was 17.5 months and the median PFS was 16.5 months. The objective intracranial response was 91% (10/11 pts) among this cohort. Notably, in the subgroup of 39 previously untreated patients, the ORR was 85% without median PFS or OS reached at the intermediate follow-up of 9.2 months. Antitumor activity of selpercatinib was observed regardless of the specific RET fusion partner. The most common severe AEs were hypertension, hepatotoxicity, hyponatremia and lymphopenia, dose reduction was warranted in 30% of patients, but only 2% discontinued selpercatinib due to a drug-related AE. Results from LIBRETTO-001 trial led to the FDA-approval of selpercatinib for patients harboring RET-positive NSCLC, in May 2020. An ongoing phase III trial (NCT04194944) [78] is evaluating selpercatinb versus platinum-based chemotherapy (CT) with or without immunotherapy (IT) in treatment-naïve patients with advanced RET-fusion positive NSCLC. The next-generation TKI pralsetinib (BLU-667), selectively developed to target RET, demonstrated meaningful preclinical activity in a wide variety of tumors with activated RET kinase [79][80]. Preliminary data from the ongoing phase 1/2 ARROW trial (NCT03037385) demonstrated potent and durable activity and tolerability of pralsetinib in the cohort of patients with advanced RET-fusion positive NSCLC [74]. Among 116 patients with RET-positive NSCLC, 80 patients had received prior platinum treatment and 26 patients were treatment-naïve: the ORRs were 61% and 73%, respectively, with a DCR of 93% in the overall population. Furthermore, ORR was similar regardless of RET fusion partner (72% of patients had KIF5B-RET fusion NSCLC, 16% had CCDC6-RET fusion NSCLC and 12% presented other fusion) or central nervous system (CNS) involvement (56%). Most treatment-related AEs were grade 1–2 and included anemia, hepatotoxicity, constipation and hypertension. Discontinuation of treatment due to side effects was reported in 4% of patients of the safety population (all tumor types) [81]. In September 2020 pralsetinib received FDA-approval for the treatment of RET-fusion positive NSCLC patients. The international randomized phase III AcceleRET Lung study (NCT04222972) [82] is currently evaluating pralsetinib compared to standard of care as first-line in RET-positive metastatic NSCLC.

